- LOGIN
- MemberShip
- 2026-07-28 19:45:23
- Policy
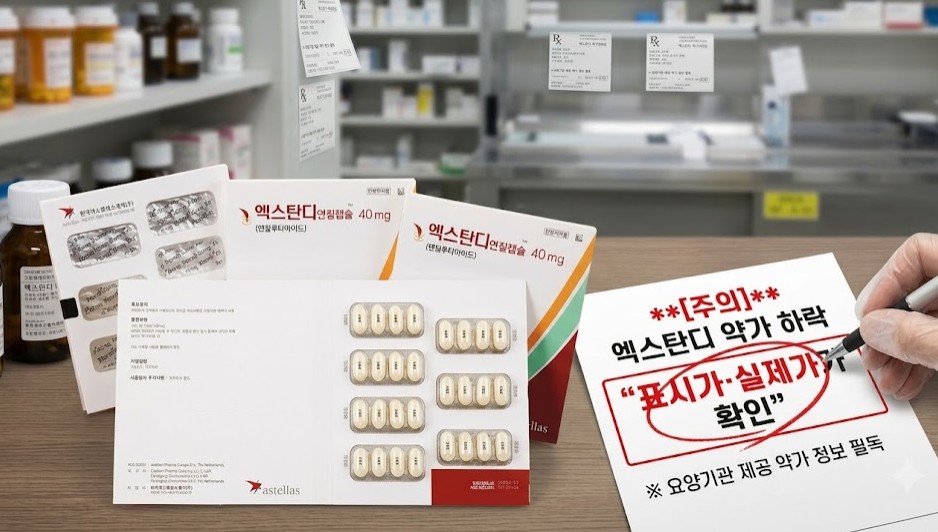
- Xtandi price to be cut following generic entry
- by Jung, Heung-Jun Jul 28, 2026 08:57am
- Reimbursement prices for eight products, including the prostate cancer treatment Xtandi and three pelubiprofen-containing analgesics, will be reduced next month.Xtandi's price will be lowered starting August 1 following the listing of generic versions in late June. Xtandi’s 70% price premium will remain in effect through June next year.However, because Xtandi is covered under Korea's Flexible Pricing Agreement system, its listed reimbursement price will remain unchanged. Healthcare providers are therefore advised to carefully verify the actual reimbursed price using the official drug price information provided to medical institutions.According to industry sources on July 27, reimbursement prices for Astella’s Xtandi Soft Cap 40 mg (enzalutamide), Xtandi Tab 40 mg, and Xtandi Tab 80 mg will be reduced next month.Among the drugs subject to price cuts next month, the actual price of Xtandi should be verified because it is covered by a flexible pricing agreement. AI-generated image.A 70% price premium was applied to the original Xtandi, with generic versions from Hanmi Pharmaceutical, Alvogen Korea, and other manufacturers listed for reimbursement on June 28 following the original drug’s patent expiry. The premium will remain in place until June 28 next year.Because the listed price differs from the actual reimbursed price, healthcare providers should refer to the official reimbursement price information available to authorized users when confirming the reduced price.Prices for three pelubiprofen products– the generic versions Yungjin Pharm's Pelps Tab, Huons' Pelroen Tab, and the salt-modified version Daewon Pharmaceutical's Pelubi S Tab (pelubiprofen tromethamine)- will also be adjusted to KRW 96. The reductions follow the Supreme Court's dismissal of a lawsuit seeking to suspend reimbursement price cuts, after which the original Pelubi Tab was subject to an administrative price reduction in May.DKSH Korea's Romiplate Inj 250 μg (romiplostim) for thrombocytopenia will be voluntarily reduced from KRW 376,881 to KRW 363,313, while the price of JW Pharmaceutical's Rabekhan Tab 10 mg (rabeprazole sodium) for gastroesophageal reflux disease will be reduced from KRW 517 to KRW 514.Meanwhile, reimbursement prices for several products will increase by more than twofold. Following review by the Drug Reimbursement Evaluation Committee, price increases have been approved for single-source medicines deemed essential for patient care that have no therapeutic alternatives or are supplied at relatively low prices.The price of the corneal preservation solution Zenith Pharm's Optisol GS will increase from KRW 152,119 to KRW 330,000. Several products from the Korea Institute of Radiological & Medical Sciences will also receive price increases, including KIRAMS Thallium Chloride (Tl-201) Inj (KRW 30,600 → KRW 34,100), KIRAMS Sodium Iodide (I-123) Solution (KRW 17,000 → KRW 19,100), KIRAMS Sodium Iodide (I-123) Injection (KRW 19,920 → KRW 27,100), KIRAMS Meta-Iodobenzylguanidine I-123 Injection (KRW 48,500 → KRW 49,100).
- Policy
- Supply halted "Keral Inj"…Huons 'first generic' wins nod
- by Lee, Tak-Sun Jul 28, 2026 08:57am
- Product photo of 'Keral Inj" A newly authorized generic injection of dexketoprofen, a nonsteroidal anti-inflammatory drug (NSAID) indicated for postoperative pain management and acute low back pain, is set to alleviate severe domestic supply disruptions in South Korea caused by the discontinuation of the original drug.According to the Ministry of Food and Drug Safety (MFDS) on the 27th, South Korea’s pharmaceutical company Huons officially secured marketing authorization for ‘Huons dexketoprofen Inj.’ The approved product is the first generic equivalent in South Korea referencing ‘Keral Inj’ (dexketoprofen trometamol). It is an innovator drug, and Hyundai Pharm holds its local commercial rights.Keral Inj is an active enantiomeric formulation that contains exclusively the S-enantiomer isolated from racemic ketoprofen, delivering rapid onset of action and potent analgesic efficacy. It has been routinely prescribed to patients requiring rapid pain relief, particularly in surgical postoperative care and acute low back pain settings.However, domestic supply of Keral Inj, imported from Italian pharmaceutical firm Menarini, was halted in January this year following the termination of its active pharmaceutical ingredient (API) supply agreement. Hyundai Pharm formally reported the supply interruption to the MFDS, citing API procurement challenges and leaving clinical sites struggling to secure alternative injectable analgesics. According to MFDS import statistics, Keral Injection recorded an import value of $129,804 (approximately KRW 180 million) in 2024.Although the underlying patents for Keral Inj expired back in November 2013, generic development had remained absent for over a decade. Huons’ successful authorization of the first-in-line generic is expected to stabilize the domestic supply chain of dexketoprofen injectables.The commercialization of Huons’ first generic is anticipated to resolve the supply-demand imbalance within the postoperative pain management market, restoring normal therapeutic options for both healthcare providers and patients.A biopharmaceutical industry source noted, “The sudden domestic supply disruption earlier this year, due to API procurement issues, created significant operational uncertainty across clinical sites, and added, “Given that Huons, a company equipped with specialized manufacturing capabilities in ampoule-packaged injectables, secured rapid authorization to commercialize this first generic, it will contribute substantially to stabilizing the supply of acute pain management therapeutics.
- Policy
- 515 days required from GIFT designation to approval
- by Lee, Tak-Sun Jul 27, 2026 08:43am
- New drugs that received marketing authorization this year (2026) through the Ministry of Food and Drug Safety's 'Global Innovative products on Fast Track (GIFT)' program took an average of 515 days from the designation date to final approval. Although the MFDS has significantly shortened its actual review time, a significant amount of time was taken by the pharmaceutical companies to prepare and supplement extensive global approval data.Dailypharm’s analysis of 10 products (GIFT Nos. 50–59) that obtained marketing approval through the MFDS' GIFT program in 2026 found that the average elapsed time from GIFT designation to final approval was 515.5 calendar days.The fastest approval was achieved by ‘Wainua Autoinjector (eplontersen),’ developed by AstraZeneca Korea for transthyretin-mediated amyloidosis. The product received marketing approval in July 2026, just 305 days after receiving GIFT designation in September 2025.In contrast, Verto Korea’s orphan drug ‘Joenja Tab’ recorded the longest timeline at 801 days, from its GIFT designation in April 2024 to final approval. Other products that took more than a year and a half included ‘Lamzede Inj’ from Kwangdong Pharm (767 days) and ‘Rimqarto Inj’ from Curocell (631 days)l, Korea's domestically developed CAR-T therapy.Time from GIFT designation to approval for key products approved in 2026The average timeline of approximately 17 months (515 days) is largely affected by the period required for companies to prepare and submit supplemental data. In other words, the entire period between GIFT designation and final approval should not be interpreted as a delay in the MFDS review process. Nevertheless, further efforts to minimize the need for additional submissions will be necessary to ensure that patients gain faster access to innovative medicines.Through the GIFT program, the MFDS aimed to shorten the statutory review period by 25%, completing reviews within 90 working days instead of the standard 120 working days. In practice, the agency's actual review time has been shortened, averaging around 60 to 70 days.The challenge arises when the MFDS requests additional information during the review process, at which point the statutory review clock stops. Industry sources noted that because many GIFT-designated products are innovative medicines from global pharmaceutical companies or advanced biopharmaceuticals, it often takes several months, or even more than a year, to coordinate GMP inspections at overseas manufacturing sites, conduct additional analyses of multinational clinical data, complete quality verification, and prepare supplementary materials in consultation with global headquarters.Building on the expertise gained through the GIFT program's ‘expedited review and rolling review’ system, the MFDS is also accelerating efforts to shorten approval timelines for all new drugs.After reducing the average review period for new drug approvals from approximately 420 days to 295 days, the agency introduced a new 240-day review framework in 2026, which is one of the fastest in the world. To support this initiative, it significantly expanded its review workforce and replaced the previous sequential review structure for nonclinical, clinical, and quality assessments with a parallel review system conducted simultaneously across departments.The MFDS has also institutionalized at least two ‘pre-submission face-to-face meetings ‘ before companies file marketing authorization applications to reduce approval delays caused by requests for additional information. By providing guidance and checklists in advance to improve documentation quality, the agency aims to minimize clock-stops during the review process.An MFDS official said, "We are fundamentally transforming the drug review system to create an environment in which patients can gain access to new medicines faster than anywhere else in the world. We will continue strengthening the predictability and transparency of the GIFT program while working closely with industry so that companies can bring innovative medicines to market without the burden of extensive supplementary submissions."

- Policy
- Differentiated R&D investment ratio applied to innovative pharma
- by Lee, Jeong-Hwan Jul 27, 2026 08:43am
- AI-generated imageThe Ministry of Health and Welfare (MOHW) is implementing the amendment to the "Enforcement Decree of the Special Act on Designation and Support of Pharmaceutical Industry," establishing differentiated annual research and development (R&D) investment thresholds for "Innovative Pharmaceutical Company" certification based on corporate revenue scale and global manufacturing capabilities.Under the revision, variable R&D investment ratios ranging from 7% to 9% will be applied using an annual revenue benchmark of KRW 100 billion. Notably, companies that have secured advanced Good Manufacturing Practice (GMP) certifications from the United States or Europe will be eligible for a lowered threshold of 5%, which is expected to ease the certification burden for drugmakers expanding into international markets. On July 24, according to biopharmaceutical industry sources, the amended Enforcement Decree, which took effect on the 21st, codifies the specific R&D expenditure requirements into three distinct categories under Article 2-2 ("Annual Research and Development Expenditure Requirements"). Differentiated 7–9% R&D sizes based on KRW 100 billion criterion…5% threshold for global GMP holdersFirst, small and medium-sized enterprises (SMEs) and mid-tier pharmaceutical companies with annual pharmaceutical revenues under KRW 100 billion must invest either at least KRW 7 billion annually or at least 9% of their annual pharmaceutical sales in R&D to meet the certification criteria.In contrast, large-scale pharmaceutical enterprises with annual pharmaceutical revenues of KRW 100 billion or more must allocate at least 7% of their annual sales to R&D to qualify for the Innovative Pharmaceutical Company designation. A notable feature of the amendment lies in the relaxed benchmark for companies equipped with global manufacturing capabilities. Pharmaceutical enterprises holding Good Manufacturing Practice approvals (such as US cGMP or EU-GMP) from regulatory authorities in the United States (FDA) or the European Union (EMA) will see their required R&D investment threshold lowered to 5% of annual pharmaceutical revenue. This provision effectively recognizes extensive investments in world-class manufacturing infrastructure as an innovative activity equivalent to direct R&D. Averaged sales from three previous fiscal year…emphasizing accounting transparencyThe amendment also clarifies the specific accounting principles and calculation methods for R&D expenditures and sales. Only revenues and expenses directly tied to "pharmaceutical products" as defined under the Pharmaceutical Affairs Act will be recognized, explicitly excluding performance metrics from non-pharmaceutical business divisions such as health functional foods or cosmetics.To prevent administrative confusion arising from short-term financial volatility, compliance will be evaluated based on the average pharmaceutical R&D expenditure and average pharmaceutical sales over the "immediately preceding three fiscal years," counting back from the fiscal year in which the company submits its certification application. For newly established companies operating for less than three years, metrics will be annualized based on performance generated up to the date of application.To enhance financial transparency, the amendment mandates that accounting for qualifying R&D expenses must strictly adhere to statutory accounting standards established under the Act on External Audit of Stock Companies. Details regarding specific expense items eligible for inclusion under pharmaceutical R&D costs will be outlined in a separate administrative notification issued by the Minister of Health and Welfare.Following the enforcement of this decree, the MOHW is expected to finalize the broader structural amendment of the Innovative Pharmaceutical Company certification system.
- Policy
- “Division of industry within the MOHW”…fostering pharma biotech
- by Lee, Jeong-Hwan Jul 24, 2026 08:21am
- Sung-il Oh, the Head of the Division of Pharmaceutical and Biotech Industry"I consider the Division of Pharmaceutical and Biotech Industry as the Division of Industry within the Ministry of Health and Welfare. While the traditional duties of the Office for Healthcare Policy on regulatory laws under the Medical Services Act and the Pharmaceutical Affairs Act, the primary mission of the Division of Pharmaceutical and Biotech Industry is fundamentally to promote and foster technology, markets, and the industry. My priority is to listen extensively and meet as frequently as possible with pharma companies, biotech firms, and healthcare sector stakeholders to build an environment where the industry can perform at its best."Core mission of the Division of Pharmaceutical and Biotech Industry under the Ministry of Health and Welfare (MOHW) include fostering domestic blockbuster novel drugs, providing policy support for the pharmaceutical and biotech sectors to address supply instabilities of essential medicines, modernizing the Innovative Pharmaceutical Company certification system, and nurturing the global pharmaceutical and biotech industry.Sung-il Oh, newly appointed as the Head of the Division of Pharmaceutical and Biotech Industry on July 22, clearly understood the aim of the division and its key responsibilities, despite returning to healthcare administration after a hiatus.Meeting with the press that day, Oh explained that while the current government, including the MOHW, considers strengthening public access to healthcare and enhancing patient convenience a major pillar of healthcare policy, it also recognizes the promotion and development of the pharmaceutical and biotech industry as a vital task to achieve.In outlining his aspirations as the new division head, Oh pledged to "listen as much as possible and meet as many people as possible." As the administration has defined the pharmaceutical and biotech industry as a key growth driver for the nation's future and signaled regulatory rationalization for its fostering and promotion, his commitment signifies an intention to directly observe and hear what regulatory improvements and policies the market actually requires.Oh highlighted, "The current government has a very keen interest in promoting the pharmaceutical and biotech industry, something I observed during my previous role as the Regulatory Reform and Legal Affairs Officer," and added, "Unlike the previous administration where the Regulatory Reform Committee functioned under the Prime Minister, the current administration's Regulatory Rationalization Committee is chaired directly by the President. Biotech has consistently been a major agenda item at presidential meetings."Oh continued, "The advice from senior officials was to listen first. I am personally experiencing that the pharmaceutical and biotech industry has many factors distinct from general healthcare administration, and I am studying various issues extensively." He stated, "I maintain an open-minded approach toward everyone who comes to visit. For now, my focus will be on properly executing well-crafted existing policies, such as the reform plan for the Innovative Pharmaceutical Company criteria."Oh concluded by stating, "Government interest in regulatory rationalization is high, and as the government believes that promoting the pharmaceutical and biotech industry will expand the nation's future growth engines, there seems to be deep deliberation at the top," and added, "While general healthcare administration primarily deals with occupational disputes and conflict mediation, and national health insurance focuses on fee adjustments, the Division of Pharmaceutical and Biotech Industry is a unit built to promote and foster. Perspective needs to shift significantly. I intend to learn thoroughly right from the basics."
- Policy
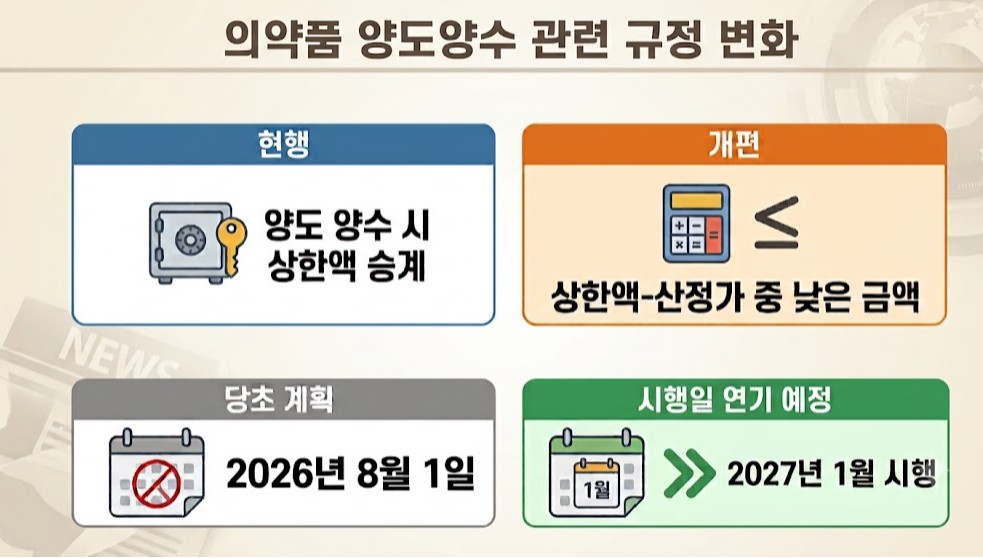
- Ban on succession of price caps deferred until next year
- by Jung, Heung-Jun Jul 24, 2026 08:21am
- The government will postpone implementation of a new rule blocking the succession of reimbursement price caps during pharmaceutical product transfers and acquisitions until next year, following a grace period through the second half of this year.The change regarding price cap succession in product transfers was not included in the drug pricing reform agenda reviewed by the Health Insurance Policy Deliberation Committee (HIPDC) in March. The government has likely decided to delay implementation in consideration of on-site acceptability.Discussions on improving the implementation of the revised drug transfer and acquisition rules took place during a public-private consultative meeting involving the Ministry of Health and Welfare, the Health Insurance Review and Assessment Service (HIRA), the National Health Insurance Service (NHIS), and industry representatives on July 22.AI-generated imageThe revision to the drug product transfer and acquisition system was included in the Ministry’s May amendment to the 'Standards for Drug Pricing Decisions and Adjustments.' Until now, for products transferred between companies, the same reimbursement price ceiling as before the transfer was applied.Under the revised system, however, products acquired through transfers will instead receive whichever is lower—the newly calculated reimbursement price or the existing price ceiling—to prevent companies from using transfers to circumvent the revised drug pricing system.Because the generic drug pricing rate will change under the new pricing framework, transferred products will effectively be subject to the newly calculated reimbursement price rather than retaining the previous reimbursement ceiling.The rules governing pharmaceutical product transfers have undergone several revisions over the years. Since 2021, however, transferred products have generally been allowed to inherit the existing reimbursement price ceiling.Because the Ministry had initially planned to implement the revised notification on Aug. 1 following its administrative notice in May, the timeline was considered too tight, prompting agreement to introduce a grace period.With implementation now postponed until January next year, products currently under transfer negotiations are expected to proceed without disruption.A wave of last-minute product transfers is expected as companies reorganize their product portfolios in the second half of the year in response to the broader drug pricing reform,Since this will be the final grace period during which acquiring companies can still inherit the existing reimbursement price ceiling, transfer activity is expected to become more active than usual.

- Policy
- Post-listing price cuts to be made every April and October
- by Jung, Heung-Jun Jul 24, 2026 08:20am
- The government is expected to exclude reimbursement expansion-related price cuts from its routine post-listing price cuts, which will take effect next year. The decision reflects concerns that patients could face delays in benefiting from lower drug costs, as well as the administrative complexity of refund procedures.At a public-private consultative meeting on drug pricing reform held on July 22, government and industry representatives discussed measures to improve the standardization of post-listing price cuts.In March, the Health Insurance Policy Deliberation Committee (HIPDC) agreed to align the timing of price cuts under various post-listing management mechanisms to April and October to improve the predictability of reimbursement price reductions.The routine post-listing price cut measure was approved by the HIPDC in March. Price cuts resulting from reimbursement expansions are expected to be excluded.Until now, reimbursement prices have been adjusted whenever a new indication was approved, or reimbursement coverage was expanded. The government sought to reduce these post-listing price cuts to two scheduled adjustments each year—in the first and second half of the year—to improve predictability for the industry.Under the original proposal, reimbursement expansions themselves would still take effect immediately. However, the resulting reimbursement price reduction was set to be deferred until the next scheduled adjustment, with pharmaceutical companies later refunding the difference.For example, if reimbursement coverage were expanded in January and the reimbursement price was therefore subject to a reduction, the amount corresponding to the delayed price cut until the April adjustment would be refunded afterward by the pharmaceutical company.However, this approach raised concerns that patients would not immediately benefit from lower drug costs at the time of reimbursement expansion.In addition, implementing the refund mechanism was expected to create a substantial and unnecessary administrative burden for both pharmaceutical companies and the National Health Insurance Service. As a result, the government has largely decided to exclude reimbursement expansion-related price cuts from the routine post-listing price adjustment schedule.Meanwhile, the government had previously planned to abolish actual transaction price-based price cuts after expanding incentives for lower-price drug purchasing but has decided to maintain the current system due to concerns about potential drawbacks. Reimbursement re-evaluations will also continue to be conducted when warranted rather than on an annual basis. The timing of price adjustments under these mechanisms will likely be brought into line with the new routine schedule.
- Policy
- Novartis to build ₩140 Billion RLT Plant in Korea
- by Lee, Jeong-Hwan Jul 23, 2026 09:08am
- The multinational pharmaceutical company Novartis will make a major investment of approximately KRW 140 billion to foster a radioligand therapy ecosystem in Korea.The investment is expected not only to establish a domestic production base for the company, but also to significantly improve access to treatment.The fact that investment in Korea by Novartis and other global pharmaceutical companies is expanding beyond clinical trials, research and development, and open innovation into the construction of manufacturing facilities is also significant.On the 21st, the Ministry of Health and Welfare (Eun-Kyeong Jeong, Minister) signed a memorandum of understanding with Novartis (Judith Love, President of Asia Pacific, Middle East and Africa) on the creation of an ecosystem for radioligand therapy in Korea at Novotel Ambassador Seoul Yongsan.Radioligand therapy combines a radioactive isotope with a ligand that binds to a target protein on cells. It is an innovative advanced biotherapeutic technology that kills cancer cells by directly breaking their DNA. One representative example is Novartis’ ‘Pluvicto Inj,’ which received approval from the Ministry of Food and Drug Safety in 2024 and has since been administered to patients in Korea.Under the agreement, Novartis plans to invest approximately KRW 140 billion to substantially strengthen Korea’s radiopharmaceutical industry capabilities.Specifically, Novartis will build an RLT manufacturing facility in Korea and establish an advanced cold-chain logistics network. It will also expand the number of hospitals capable of administering RLT from the current 10 to 30.Furthermore, the company will focus investment in building the broader RLT ecosystem, including the training of domestic specialist researchers to global standards.Novartis has invested in and collaborated with Korean pharmaceutical companies in various areas, including technology transfer and clinical research. Its latest investment in an RLT production facility, which expands these collaborative achievements into the advanced biopharmaceutical field, is expected to strengthen Korea’s research, development and manufacturing capabilities in radiopharmaceuticals.Minister Jeong said, “We sincerely welcome Novartis's decision to make a large-scale manufacturing investment in Korea. It is meaningful that a global leader in the pharmaceutical and biotechnology market has shown confidence in Korea's outstanding technological capabilities and potential. This agreement will serve as a significant turning point in providing innovative treatment options to patients in Korea.”Judith Love, President of Novartis APMA, added, “This agreement represents an important step in accelerating the advancement of Korea’s RLT ecosystem and strengthening the country’s role in future healthcare innovation. We will work closely with the Korean government and healthcare stakeholders to benefit more patients with our innovative cancer treatments.”
- Policy
- Lilly’s 'Cymbalta' is withdrawing from the KOR mkt after 20 years
- by Lee, Tak-Sun Jul 22, 2026 08:50am
- Product photo of CymbaltaEli Lilly Korea's central nervous system (CNS) therapy, 'Cymbalta Capsules' (duloxetine hydrochloride), is officially withdrawing from the South Korean market, nearly 20 years after its domestic launch.According to the Ministry of Food and Drug Safety (MFDS), Eli Lilly Korea voluntarily withdrew the product approval for Cymbalta Capsules as of July 21. This license revocation appears to be a voluntary step in line with Lilly's transition in its global business strategy.Cymbalta is the leading therapeutic in the serotonin-norepinephrine reuptake inhibitor (SNRI) class. It acts by inhibiting the reuptake of serotonin and norepinephrine, key neurotransmitters in the brain, to restore chemical balance.Although widely known as a treatment for major depressive disorder (MDD) and generalized anxiety disorder (GAD), Cymbalta also demonstrates exceptional efficacy in modulating physical pain signals, leading to its widespread prescription across various conditions, including diabetic peripheral neuropathic pain, fibromyalgia, and osteoarthritis pain.Industry analysis suggests that this withdrawal was an expected outcome. In early 2024, Eli Lilly Korea terminated its long-standing co-promotion (joint sales) agreement with Boryung for Cymbalta.Following the termination of joint sales, operational and distribution gaps, alongside recurring supply shortages of Cymbalta in the domestic market over recent years, resulting in inconvenience for healthcare professionals and patients.Portfolio adjustments at the global level also appear to have contributed. Eli Lilly Korea had previously phased out CNS drug lineups, including the ADHD medication 'Strattera' and the antidepressant 'Prozac'. Additionally, the company previously executed market withdrawals of Cymbalta in certain overseas markets, such as Australia, citing declining profitability and intensifying generic competition.While the original drug is exiting the South Korean market, patient care is not expected to be significantly disrupted. This is because the substance patent for Cymbalta expired in 2014, allowing numerous domestic generics to establish a firm presence in the market.Currently, 39 generic products (active ingredient: duloxetine) hold regulatory approval in South Korea, including those from Myung In Pharm, Whan In Pharm, Hanlim Pharm, and Daewoong Pharmaceutical. With abundant market supply of therapeutic alternatives with identical active ingredients and dosages, patients currently taking Cymbalta are expected to transition smoothly to generic formulations through consultations with their healthcare providers.
- Policy
- Bipartisan-approved bills to automatically reach plenary
- by Lee, Jeong-Hwan Jul 22, 2026 08:50am
- In-soon Nam, Vice Speaker of the 22nd National AssemblyA long-standing parliamentary practice in which bills approved by both the relevant standing committee and the Legislation and Judiciary Committee (LJC) are delayed in the plenary session without clear justification could soon come to an end.Legislation is being proposed to require that any bill approved by agreement between the Legislation and Judiciary Committee chair and floor leaders of both the ruling and opposition parties be automatically placed on the plenary agenda if 45 days have passed since its approval by the committee.The bill also requires standing committees to complete their review of bills jointly introduced by lawmakers from both parties within 30 days for partial amendments and within 45 days from referral for newly enacted or fully revised legislation.In particular, the proposal would prohibit filibusters on bills approved by consensus between the committee chair and floor leaders of both parties in either the relevant standing committee or the Legislation and Judiciary Committee.On July 21, National Assembly Vice Speaker In-soon Nam of the Democratic Party announced that she had introduced the partial revision to the National Assembly Act.The core provision would require bills that have passed both the relevant standing committee and the Legislation and Judiciary Committee through bipartisan agreement to be automatically referred to the plenary session after 45 days.The proposal fulfills one of Nam's pledges to improve parliamentary procedures by preventing bipartisan bills from being indefinitely delayed before reaching the plenary session without valid justification.If enacted, it would help prevent situations in which bipartisan bills remain stalled for over half a year despite clearing all committee stages, such as legislation prohibiting telemedicine platform operators from establishing or operating pharmaceutical wholesalers and extending dual penalties for illegal rebates to match those already applied to physicians and pharmacists.Nam said the proposal is intended to improve the predictability of plenary schedules while ensuring the timely review of livelihood-related legislation.A key provision of the proposed amendment is the mandatory referral of bills approved through bipartisan agreement to the plenary session. Specifically, bills approved by agreement between the chair of the Legislation and Judiciary Committee and the floor leaders of each negotiating party would be required to be placed on the plenary agenda once 45 days have elapsed after committee approval.The bill also includes measures to prevent abuse of filibusters by prohibiting them for bills passed through bipartisan agreement in either the relevant standing committee or the Legislation and Judiciary Committee, thereby preventing politically motivated delays to livelihood legislation.Another provision creates a legislative fast-track for bills jointly introduced by lawmakers from different negotiating parties, that is, the ruling and opposition parties. Standing committees would be required to review partial amendment bills within 30 days of referral, and newly enacted or fully revised bills within 45 days.To improve the predictability of parliamentary proceedings, the proposal would also require the Speaker to announce and publish the day's plenary agenda at least 24 hours before the session convenes whenever the agenda is finalized on the same day.The proposed amendment is expected to serve as a strong institutional safeguard to create a more effective National Assembly and ensure that at least bills agreed upon by both parties are not derailed by political disputes and can take effect in a timely manner.Nam said, "An amendment was long overdue, with current rules continuing to be abused, with filibusters being requested for political reasons even on bills agreed upon by both parties. The amendment is intended to prevent bipartisan bills from being delayed for extended periods in the Legislation and Judiciary Committee or the plenary session, and to avoid repeated uncertainty in parliamentary scheduling caused by political confrontation."