- LOGIN
- MemberShip
- 2026-07-29 16:11:11
- Company
- Organon commences exclusive sales of Atozet and Vytorin
- by Kim, Jin-Gu Jul 29, 2026 08:50am
- Product photo of ‘Atozet’ and ‘Vytorin’Organon Korea and Chong Kun Dang Pharmaceutical are terminating their co-promotion agreement for the dyslipidemia therapies 'Atozet' and 'Vytorin'. According to pharmaceutical industry sources on the 28th, Organon Korea recently announced on its internal notice board that, following strategic business discussions with Chong Kun Dang, the two companies agreed to terminate their co-promotion agreement for Atozet and Vytorin, effective July 31. Consequently, starting August 1, Organon Korea will be responsible for all distribution, sales, and marketing operations for both products.Atozet and Vytorin are ezetimibe-based lipid-lowering combination drugs. Atozet is a combination therapy containing ezetimibe with atorvastatin, while Vytorin combines ezetimibe with simvastatin. Both therapies work by a dual-inhibition mechanism that simultaneously suppresses cholesterol absorption in the intestines and cholesterol synthesis in the liver. The partnership between the two companies commenced in January 2016 through a co-promotion agreement between MSD Korea and Chong Kun Dang. Following Organon's spinoff from MSD in 2021, the partnership was maintained, continuing their collaboration for a full decade.The collaboration between Chong Kun Dang’s strong primary care clinic sales network and MSD/Organon’s original branding yielded synergy, establishing Atozet as a blockbuster therapy generating over KRW 100 billion in annual prescription sales. The termination of the agreement is expected to have a different impact on the future sales strategies, marketing operations, and financial performance of both firms. Organon Korea, transitioning to a direct, sole-commercialization model, is expected to maximize product profit margins. The company aims to improve profitability after eliminating co-promotion commission payments previously disbursed to Chong Kun Dang.However, a key challenge for Organon Korea will be whether its internal sales department can effectively bridge the gap left by Chong Kun Dang’s extensive primary care clinic sales infrastructure. Given that the dyslipidemia market heavily relies on clinic-level prescriptions, defending market share during the initial transition to direct sales will be the primary goal.Quarterly prescription sales for ‘Atozet’ and ‘Vytorin’ (unit: KRW 100 million, source: UBIST). GREEN-Atozet, RED-VytroinConversely, Chong Kun Dang is expected to transition its commercial focus toward its proprietary generic version, 'Lipilouzet'. Chong Kun Dang secured regulatory approval for Lipilouzet in October 2020 and currently serves as a contract manufacturing organization (CMO), producing the same generic formulation for 19 other pharmaceutical companies. While co-promoting a major original blockbuster drug helps sustain overall sales scale, sharing margin cuts reduces profitability. In contrast, commercializing its own generic, Lipilouzet, alongside generating CMO revenues from third-party partners, offers significantly higher profit margins. Thus, while Atozet's departure will temporarily reduce Chong Kun Dang's sales volume, the company anticipates improved overall operating margins, driven by a higher proportion of high-margin products.According to the pharmaceutical market research firm UBIST, Atozet recorded KRW 66.9 billion in prescription sales during the first half of this year, marking a 7% year-over-year (YoY) increase from the KRW 62.5 billion sales in H1 of last year. Over the same period, Vytorin’s prescription sales dropped 16% from KRW 6.0 billion to KRW 5.0 billion, while Chong Kun Dang’s Lipilouzet showed a 35% YoY surge, an increase from KRW 1.5 billion to KRW 2.0 billion.

- Company
- "RSV prevention option for infants has been added"
- by Son, Hyung Min Jul 29, 2026 08:50am
- A new treatment option has entered South Korea's infant respiratory syncytial virus (RSV) prophylaxis market.Following domestic regulatory authorization of MSD Korea's long-acting antibody injection 'Enflonsia (clesrovimab)', competition in the infant RSV market is set to intensify alongside Sanofi and AstraZeneca's 'Beyfortus (nirsevimab)'.On the 29th, MSD Korea hosted a press conference at The Plaza Hotel in Jung-gu, Seoul, to celebrate the domestic approval of Enflonsia.Professor Ki Wook Yoon of the Department of Pediatrics at Seoul National University College of MedicineEnflonsia was approved in South Korea on the 1st of last month as a long-acting preventive monoclonal antibody indicated for the prevention of RSV-induced lower respiratory tract disease (LRTD) in newborns and infants. The antibody is distinguished by its fixed-dose administration design, which requires no weight-based dosage adjustments. Providing sustained protective efficacy for 5 to 6 months post-injection, a single dose delivers comprehensive coverage throughout an entire RSV epidemic season.In South Korea, RSV typically peaks between October and March. Enflonsia can be administered immediately at birth for newborns during the active season, or as a single dose before the start of the first RSV season for infants born during off-season months. RSV infections initially present with mild, cold-like symptoms. The infections frequently progress to severe lower respiratory tract complications, such as acute bronchiolitis and pneumonia, in infants under one year of age, often requiring emergency intervention and inpatient hospitalization.Professor Ki Wook Yoon of the Department of Pediatrics at Seoul National University College of Medicine (Public Relations Director of the Korean Society of Pediatric Infectious Diseases) stated, "Young infants are exceptionally vulnerable to severe RSV disease due to their immature immune systems and narrow airway anatomy," and added, "When infection progresses to severe lower respiratory disease requiring medical management, the clinical burden centers heavily on acute hospitalizations."Professor Yoon added, "Infant RSV prophylaxis strategies must expand beyond simply blocking initial transmission to substantially mitigating the burdens experienced by pediatric patients and their families, including acute hospital admissions and severe lower respiratory tract illness."According to a nationwide Korean study (2007–2019) published last year, which analyzed National Health Insurance (NHI) claims data, 44.7% of pediatric RSV cases in South Korea under age 5 required hospitalization.Infants aged 6 to 11 months were the most heavily impacted cohort, accounting for 48.2% of total RSV hospitalizations and 57.3% of intensive care unit (ICU) admissions among children under 5. Infants under 6 months of age demonstrated the longest average hospital stay at approximately 8.35 days.In the Phase 2b/3 CLEVER clinical trial, a single dose of Enflonsia reduced the incidence of RSV-associated medically attended lower respiratory tract infections by 60.5%. It decreased the risk of RSV-associated hospitalizations by 84.3%.Furthermore, in the Phase 3 SMART trial evaluating high-risk infants and young children, Enflonsia demonstrated a safety profile comparable to the existing standard-of-care RSV antibody, 'Synagis (palivizumab)', confirming consistent preventive efficacy.Professor Yoon noted, "Because the burden of RSV hospitalizations and severe lower respiratory tract disease concentrates heavily during an infant's first year of life, evaluation of preventive interventions must consider how effectively they reduce medically attended severe disease and hospital admissions alongside overall infection rates."Professor Yoon concluded by emphasizing, "Given that Enflonsia demonstrated efficacy in reducing RSV-related lower respiratory infections as well as hospitalizations, the evidence demonstrates the clinical value of this drug as an essential preventive option capable of alleviating the overall disease burden for infants entering their first RSV season."

- Company
- Pharma companies accelerate investment in future growth
- by Cha, Ji-Hyun Jul 29, 2026 08:49am
- Major Korean pharmaceutical and biotechnology companies are moving to expand production and research facilities. Their investments range from trillion-won biologics manufacturing plants to large-scale research complexes. The strategy is to use accumulated cash-generation capacity and financial headroom to expand production and prepare proactively for future demand.According to the Financial Supervisory Service on the 28th, GC Biopharma recently decided to invest KRW 140 billion in a new production line at its Ochang plant in North Chungcheong Province. The investment is equivalent to 10% of the company’s consolidated equity as of the end of last year and will run through the end of 2030.The project is intended to establish a production base for a subcutaneous formulation of the immunoglobulin therapy Alyglo. Alyglo is a plasma-derived product manufactured by isolating and purifying immunoglobulin from blood plasma and is used to treat primary immunodeficiency diseases, including congenital immunodeficiency. GC Biopharma is developing a subcutaneous version of Alyglo, which is currently administered intravenously, to improve convenience for patients. The company aims to submit a Phase III clinical trial application to the US Food and Drug Administration next year. The new line is intended to secure production capacity in advance of commercialization and anticipated growth of US demand.Ahn-Gook Pharm held a board meeting on the 14th and approved a KRW 48.5 billion investment to expand its Hyangnam plant in Hwaseong, Gyeonggi Province. The amount is equivalent to 29% of consolidated equity as of the end of last year.The investment will be made in the plant located in the pharmaceutical industrial complex in Hyangnam-eup, Hwaseong. Construction began that day and is scheduled for completion by the end of December next year. The company said the expansion is being made to increase production capacity.Chong Kun Dang will build a biopharmaceutical research complex in the Baegot district of Siheung, Gyeonggi Province. The facility investment totals KRW 392.5 billion, equivalent to 39% of consolidated equity as of the end of last year. The company will continue investment through the end of August 2028.The project covers a 79,790.8㎡ site near 302 Baegot-dong, Siheung. Chong Kun Dang previously acquired the site for KRW 94.9 billion in June last year. The company plans to create an integrated research complex comprising biopharmaceutical research facilities, a research support center and R&D validation facilities.The new complex will conduct research and development of next-generation therapies, including cell and gene therapies, antibody-drug conjugates, recombinant proteins and bispecific antibodies. Chong Kun Dang also plans to establish an AI-enabled drug discovery platform and develop the site into an R&D hub linking candidate discovery, research, development and validation in one location. Collaboration is also expected with the nearby Seoul National University Hospital in Siheung Baegot, as well as companies and research institutes.These are not the only companies undertaking large-scale facility investments. Pharmaceutical and biotechnology companies have announced a series of investments in production and research facilities this year.HK inno.N will invest KRW 97 billion to build a new solid oral dosage manufacturing facility with a total floor area of 12,561.98㎡ on the remaining land at its Osong plant in North Chungcheong Province. The investment will continue through the end of January 2028. The company plans to secure medium- and long-term growth engines and strengthen competitiveness by expanding production capacity.Celltrion will invest KRW 1.2265 trillion to build its fourth and fifth plants at its Songdo campus in Incheon. It plans to apply automation systems and smart factory technologies to the new facilities to improve production efficiency and process flexibility. The plants will be designed to accommodate production ranging from small-volume, multi-product manufacturing to large-scale commercial output. They will be used not only for established flagship products but also for follow-on biosimilars and new drug pipelines.Celltrion is also expanding both drug substance (DS) and drug product (DP) manufacturing capacity in the US and Korea. At its DS plant in Branchburg, New Jersey, the company has increased the scale of the planned expansion to add 75,000 liters of capacity. Once completed, capacity will rise from 66,000 liters to over 141,000 liters. When the fourth and fifth Songdo plants are also completed, Celltrion’s total DS production capacity will increase from 316,000 liters to 571,000 liters.In the DP segment, a new plant at the Songdo campus is scheduled for completion this year. The facility will be able to produce 6.5 million liquid vials annually. Combined with the existing production line at Plant 2, annual capacity in Songdo will rise to 10.5 million vials. Celltrion has also selected a site for a new DP plant in the Yesan Industrial Complex in South Chungcheong Province and plans to begin design work this year. The group expects to be able to manufacture approximately 90% of global DP demand in-house once Celltrion Pharm completes the expansion of its prefilled syringe production facilities.BTGEN, formerly STgen Bio and a subsidiary of Dong-A Socio Holdings, will also invest KRW 109 billion to expand its biologics manufacturing facilities. The investment includes the expansion of buildings and production equipment and will continue through the end of March 2028. BTGEN plans to increase its biologics manufacturing capacity through the project.Industry observers attribute this acceleration in future investment to the back of strong earnings made by Korean pharmaceutical and biotechnology companies.Celltrion posted consolidated revenue of KRW 1.3937 trillion and operating profit of KRW 451.8 billion in Q2, up 45% and 86%, respectively, year over year. In addition to solid sales of existing flagship products, increased revenue from higher-margin new biosimilars such as Remsima SC and Yuflyma drove the company’s earnings growth.Profitability also improved significantly as the rising contribution of new products coincided with the depletion of high-cost inventory and better production yields. 1H operating profit rose 97% year over year to KRW 773.7 billion, while the operating margin increased by 9 percentage points to 31% from 22% a year earlier.HK inno.N is also maintaining earnings growth on the back of strong prescription drug sales. Q1 revenue rose 5% year over year to KRW 258.7 billion, while operating profit increased 31% to KRW 33.2 billion. The improvement was driven by broad growth across its prescription drug business, including IV solutions, cardiovascular drugs and oncology products. Q1 IV solution sales rose 11% year over year to KRW 37.1 billion, while cardiovascular sales increased 10% to KRW 73 billion. Oncology sales climbed 34% to KRW 29.2 billion with the addition of the in-licensed drug Avastin.Ahn-Gook Pharm also expanded its investment capacity on the back of improved earnings. Consolidated Q1 revenue increased 31% year over year to KRW 98.9 billion, while operating profit surged 174% to KRW 16 billion. Its operating margin stood at 16.2%. The company’s earnings improvement was driven by simultaneous growth in its flagship prescription drug and health and beauty businesses.Chong Kun Dang’s consolidated Q1 revenue increased 12% year over year to KRW 447.8 billion, while operating profit rose 13% to KRW 14.1 billion. GC Biopharma’s consolidated Q1 revenue increased 14% to KRW 435.5 billion, while operating profit rose 47% to KRW 11.7 billion.BTGEN is also among the companies that have posted rapid earnings growth. Its revenue increased 76% year over year to KRW 103.7 billion last year, while operating profit surged 318% to KRW 7.1 billion. Basically, the company’s revenue nearly doubled and operating profit more than quadrupled in one year. Increased global commercial production of the Stelara biosimilar Imuldosa and higher exports of an anemia treatment biosimilar drove the company’s growth. However, in Q2 this year, revenue fell 45% year over year to KRW 13.7 billion and operating profit declined 90% to KRW 400 million, with orders from major customers concentrated in 2H.The sector is regarded as establishing a virtuous cycle in which funds generated through stronger earnings are being reinvested in production and research facilities. The companies aim to use expanded capacity to meet future demand for biologics and prescription drugs and convert that demand into further revenue and profit growth. Expectations are growing that their large-scale expansion projects will help them capture future demand and further strengthen their medium- and long-term growth foundations.
- Company
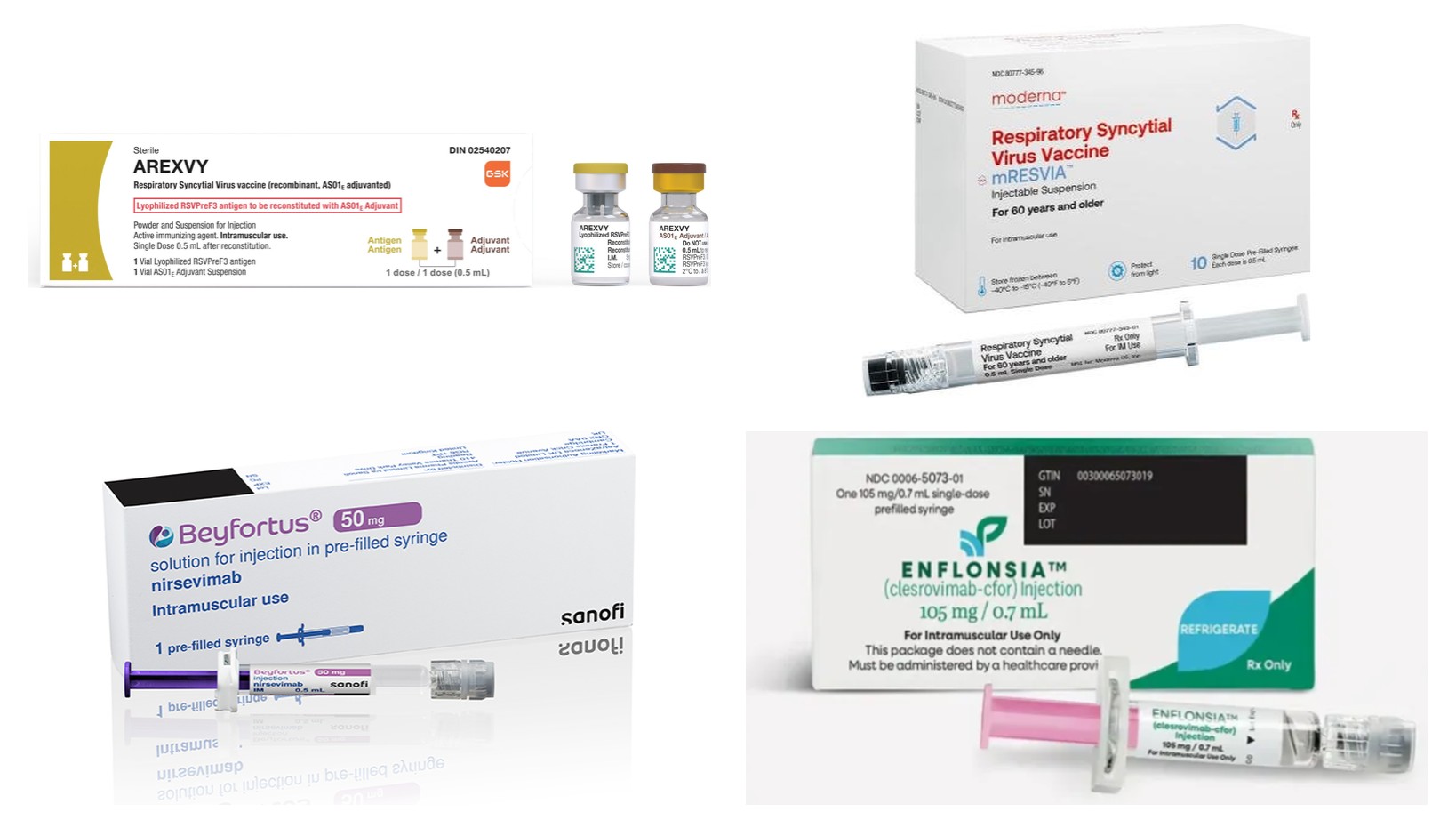
- RSV market competition intensifies in Korea
- by Son, Hyung Min Jul 29, 2026 08:49am
- The respiratory syncytial virus (RSV) prevention market is rapidly expanding across both adult and infant populations. In adults, GSK and Moderna are competing with vaccines, while in infants, Sanofi and MSD are competing with long-acting preventive antibody injections, with Pfizer’s maternal immunization vaccine also expected to enter the market.With RSV prevention options becoming more diverse, calls are growing for financial support through inclusion in the National Immunization Program (NIP) or National Health Insurance reimbursement to enable broader market uptake. In particular, because preventive antibody injections for infants are medicines rather than vaccines, policymakers still need to determine whether their access should be expanded through the NIP or health insurance reimbursement.According to industry sources on the 29th, competition is rapidly taking shape in Korea’s RSV prevention market, especially around adult vaccines and preventive antibody injections for infants.In the adult market, GSK’s Arexvy is competing with Moderna’s mRESVIA. In the infant market, MSD Korea’s Enflonsia (clesrovimab) has received domestic authorization following Sanofi’s Beyfortus (nirsevimab). Pfizer’s RSV vaccine for pregnant women, Abrysvo, is also reportedly nearing approval in Korea.RSV generally causes mild cold-like symptoms, but in infants, older adults, and patients with chronic diseases, it can progress to lower respiratory tract diseases such as bronchiolitis or pneumonia. Infants entering their first RSV season are particularly vulnerable because their immune systems are not fully developed and their airways are narrow, increasing the risk of severe disease and hospitalization.A study analyzing Korean National Health Insurance claims data found that as many as 18,434 RSV cases occurred annually among children under five from 2007 to 2019, and 44.7% of all patients were hospitalized.Infants aged 6 to 11 months accounted for 48.2% of RSV hospitalizations and 57.3% of intensive care unit admissions. Patients under 6 months had the longest average hospitalized days at 8.35 days, indicating a burden not only on infant health but also on caregiver responsibilities and healthcare resources.Indication for GSK’s Arexvy expands to high-risk adults…builds long-term protection evidenceGSK’s Arexvy, Moderna’s mRESVIAIn the adult RSV vaccine market, the expansion of Arexvy’s indication is expected to reshape the competitive landscape.GSK recently expanded the indication for Arexvy to high-risk adults aged 18 to 49. In addition to adults aged 60 and older and high-risk adults aged 50 to 59, the vaccine now covers adults aged 18 and older at increased risk of severe RSV due to chronic respiratory disease, cardiovascular disease, diabetes, chronic kidney disease, or immunocompromising conditions.The expanded indication was based on a global Phase IIIb trial in high-risk adults aged 18 to 49. The study compared immune responses in this population with those in adults aged 60 and older and demonstrated non-inferiority. The safety profile was also comparable to that observed in previously vaccinated groups.Arexvy has also generated evidence of efficacy and long-term protection in older adults. In the Phase III AReSVi-006 trial, its efficacy against RSV-associated lower respiratory tract disease during the first RSV season was 82.6% among adults aged 60 and older. The prevention effect was 94.6% among adults with at least one underlying condition.Long-term follow-up showed cumulative efficacy of 62.9% over three RSV seasons after a single dose, while efficacy against severe RSV-associated lower respiratory tract disease was 67.4%. Although efficacy declined over time, the vaccine demonstrated protection across 3 seasons without a booster dose.Moderna entered the competition with mRESVIA, which uses an mRNA platform. The vaccine is designed to produce the RSV F protein and is differentiated by extending the mRNA platform technology accumulated through COVID-19 vaccines to RSV prevention.In Korea, mRESVIA is approved for preventing RSV-associated lower respiratory tract disease in adults aged 60 and older. Whether the indication will be expanded beyond older adults to younger high-risk groups, such as chronic disease and immunocompromised patients, is expected to be a key variable in the adult market.Sanofi and MSD compete with preventive antibody injections…highlights reductions in hospitalization and severe diseaseIn the infant market, long-acting monoclonal antibodies formed the competitive landscape rather than vaccines.Sanofi’s Beyfortus is a preventive antibody injection that directly administers antibodies to newborns and infants entering their first RSV season, providing protection immediately after infection. A single dose is designed to maintain protection for approximately 5 months.RSV preventive antibody injections Beyfortus, EnflonsiaBeyfortus can be used not only in infants during their first RSV season but also in children under 24 months who remain at high risk of severe disease during their second season. The dose during the first season varies according to body weight, while a separate dose is used for high-risk children during the second season.MSD Korea entered the infant RSV preventive antibody market after receiving domestic approval for Enflonsia in June.Enflonsia is approved to prevent RSV-associated lower respiratory tract disease in newborns and infants entering their first RSV season or born during the season. It can be administered once at a fixed dose of 105 mg regardless of body weight, with protection lasting for at least 5-6 months.The global Phase IIb/III CLEVER trial supporting approval enrolled approximately 3,600 healthy preterm and full-term infants born at 29 weeks of gestation or later across 22 countries, including Korea.The study showed that Enflonsia reduced the risk of medically attended RSV-associated lower respiratory tract infection by 60.4% versus placebo through 150 days after administration. During the same period, it reduced the risk of RSV-related hospitalization by 84.2%. Through 180 days, it lowered the risk of medically attended severe RSV-associated lower respiratory tract infection by 91.7%.In the safety analysis, more than 96% of reported adverse events were mild or moderate.In the Phase III SMART trial involving preterm infants and infants with chronic lung disease or congenital heart disease who were at high risk of severe RSV, Enflonsia showed a generally similar safety profile to the established preventive antibody injection Synagis (palivizumab).Enflonsia’s fixed-dose regimen is expected to simplify administration in clinical practice as there is no need to calculate the dose based on body weight or prepare different product presentations; it may improve convenience at healthcare institutions treating infants of varying birth dates, lunar ages and weights.MSD Korea is also seeking to expand Enflonsia’s indication beyond the first RSV season to include children at high risk of severe disease during their second season.The company has obtained follow-up data from the Phase III SMART trial in children under 2 years of age who remained at high risk across two RSV seasons. Among children given Enflonsia during their second season, blood drug concentrations were similar to those observed in healthy infants during their first season, while the safety profile remained consistent with earlier results.If the indication is expanded to high-risk infants in their second season, the scope of competition with Beyfortus, which already holds that indication, is also expected to widen.Pfizer’s Abrysvo nears approval… adding maternal immunization strategyRSV vaccine AbrysvoPfizer is preparing to enter the infant RSV prevention market with a maternal immunization strategy that differs from those of preventive antibody injections.Abrysvo is administered to pregnant women so that antibodies generated by vaccination are transferred to the fetus through the placenta. Unlike preventive antibody injections administered directly to infants after birth, the vaccine is designed to provide passive immunity against RSV from birth through vaccination during pregnancy.In the US, Abrysvo was approved in 2023 for administration to pregnant women at 32 to 36 weeks of gestation to prevent RSV-associated lower respiratory tract disease and severe lower respiratory tract disease in infants through six months of age. It was the first vaccine approved to prevent RSV in infants through maternal immunization.In the Phase III MATISSE trial supporting approval, vaccination during pregnancy reduced the risk of severe RSV-associated lower respiratory tract disease in infants by 81.8% through 90 days after birth. Efficacy through 180 days was 69.4%.Abrysvo is also expected to receive approval in Korea soon. Once approved, infant RSV prevention will be divided between direct administration of a long-acting antibody after birth and transfer of antibodies before birth through maternal vaccination.Because the 2 strategies differ in their target populations and timing of administration, they are likely to be selected based on maternal vaccination status, the infant’s birth timing, underlying conditions and risk of severe disease rather than serving as complete substitutes.Prevention options become available… NIP and insurance coverage remain market variablesAs RSV prevention products become available across adult and infant populations, attention is shifting to their funding mechanisms.Given Korea’s transition to a super-aged society and the burden of hospitalization and mortality among older adults, the possible inclusion of RSV vaccines for older adults in the NIP may eventually be discussed. However, additional policy evidence is needed regarding the domestic disease burden, cost-effectiveness, and priority vaccination groups.The framework for preventive antibody injections in infants is more complex. Beyfortus and Enflonsia are used to prevent infection but are monoclonal antibodies rather than vaccines. Policymakers must therefore determine whether they should be included in the NIP for government purchase and supply or covered through health insurance and prescribed by healthcare institutions.The US Centers for Disease Control and Prevention’s Advisory Committee on Immunization Practices recommends long-acting preventive antibody injections for infants under 8 months who are entering or are already in their first RSV season. Overseas, preventive antibodies are increasingly being incorporated into public health prevention strategies.At a press conference held on the 28th to mark Enflonsia’s approval, MSD Korea also emphasized the need to secure access through either NIP inclusion or health insurance coverage.Discussions on institutional access in Korea’s RSV prevention market may therefore initially progress more rapidly in the infant segment. The government has historically prioritized the pediatric NIP over vaccination programs for older adults.With the hospitalization and severe disease burden of RSV in infants already established and multiple products demonstrating preventive efficacy, calls for financial support are increasing. Sanofi and MSD are exploring potential NIP inclusion or health insurance coverage for their preventive antibodies. Once Prizer receives approval for Abrysvo in Korea, policy discussions on infant RSV prevention are expected to broaden to include maternal immunization.
- Company
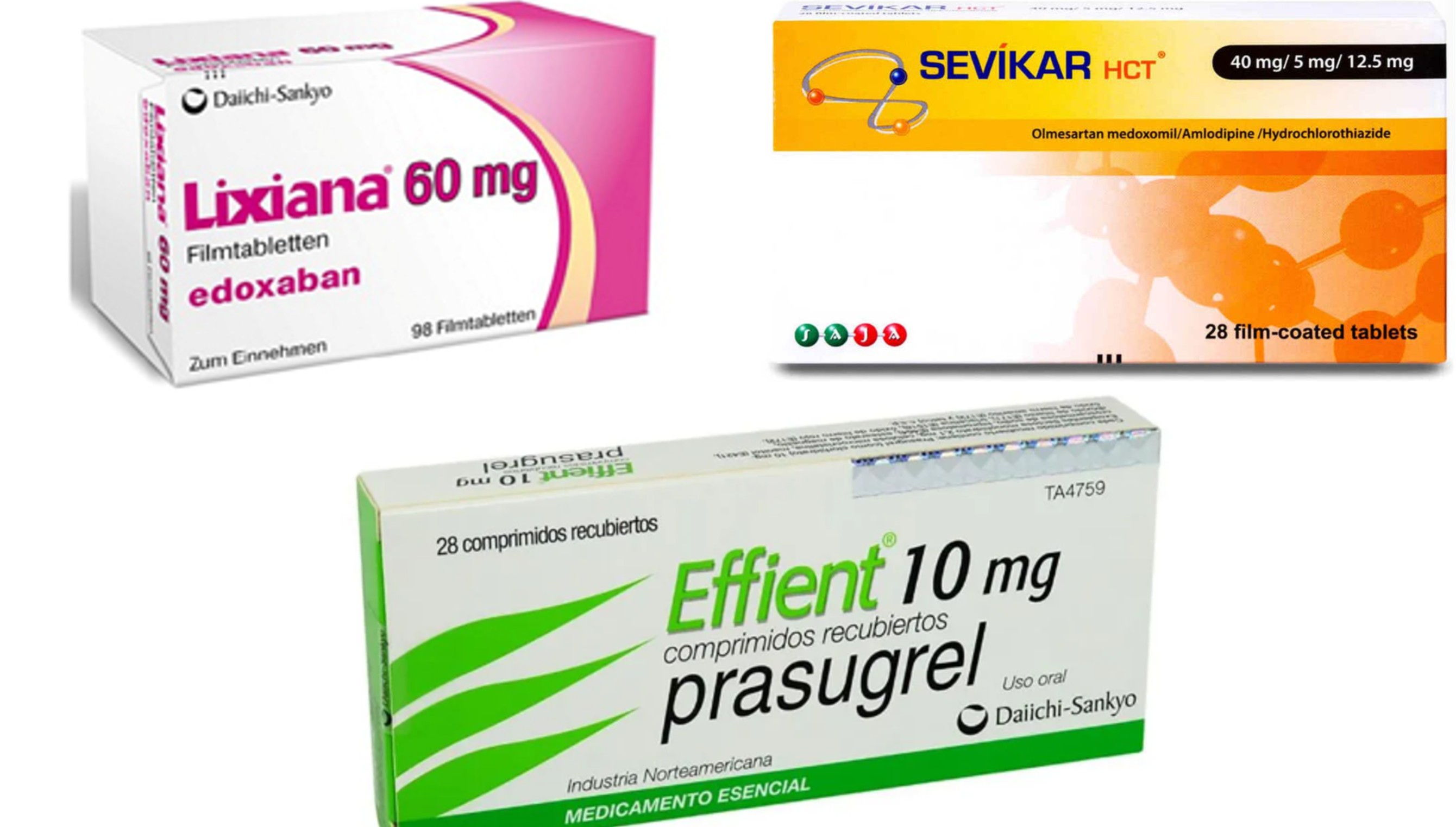
- Daiichi Sankyo's major cardiovascular drugs hit by supply shortages
- by Son, Hyung Min Jul 28, 2026 12:01pm
- Supply shortages of Daiichi Sankyo Korea’s major cardiovascular drugs are placing increasing pressure on medical sites.Production and shipment schedules have been delayed due to maintenance work at the company’s overseas manufacturing facility, resulting in simultaneous supply disruptions for the anticoagulant ‘Lixiana (edoxaban),’ the triple-combination antihypertensive ‘Sevikar HCT (olmesartan/amlodipine/hydrochlorothiazide),’ and the antiplatelet agent Efient (prasugrel).Although severity varies by product and strength, supplies of some products have deteriorated to the point that inventories are scarce across distribution channels. Hospitals, clinics, and pharmacies have reportedly been checking remaining stock or considering alternative therapies in an effort to maintain existing prescriptions.According to industry sources on July 28, supply shortages of Lixiana, Sevikar HCT, and Efient have persisted since last month.(Clockwise from the left) Lixiana, Sevikar HCT, and EfientDaiichi Sankyo Korea attributed the disruption to maintenance work at its manufacturing facility in Germany, which delayed production and shipments. The manufacturing issue is understood to be affecting product supplies not only in Korea but also across global markets.A Daiichi Sankyo Korea official said, "The products are not completely out of stock, but we have been unable to distribute enough inventory to meet market demand. However, we assess that the supply issues are gradually easing."Lixiana is currently experiencing the most severe supply shortage. Inventories of Lixiana 15mg and 30mg have virtually been depleted, and it is understood that even 60mg, which had relatively remaining stock, is left with almost no supply capacity.Some healthcare institutions are receiving limited quantities that can only meet individual patient prescriptions. Even major wholesalers and the distribution network of Daewoong Pharmaceutical, which co-promotes the product, have been unable to secure sufficient inventories, making it difficult to fulfill orders from medical institutions.Lixiana is a direct oral anticoagulant (DOAC) indicated to reduce the risk of stroke and systemic embolism in patients with non-valvular atrial fibrillation and to treat deep vein thrombosis and pulmonary embolism. Because anticoagulants are often taken continuously over the long term, supply interruptions may force physicians to switch patients to alternative products within the same class.Sevikar HCT and Efient are also experiencing varying degrees of shortages depending on the strength, but overall, supplies remain insufficient to fully meet demand.Sevikar HCT is an olmesartan-based triple-combination antihypertensive, while Efient is an antiplatelet therapy used to prevent thrombotic cardiovascular events in patients with acute coronary syndrome.Because all three products are maintenance therapies for patients with cardiovascular disease, prolonged supply disruptions could further increase the burden on-site. Even when alternative products or therapies are available, treatment changes must be made carefully based on each patient's clinical condition and response to previous therapy.A distribution industry official said, "Lixiana 15 mg and 30 mg are virtually impossible to obtain, and inventories of the 60 mg strength are also running very low. Even when hospitals place orders, they are often supplied with only part of the requested quantity or with only the minimum amount needed for individual patients."The official added, "Neither wholesalers nor the co-promotion partner have sufficient inventories, suggesting that this is not a problem at a particular distribution stage but rather an overall product supply shortage. The disruption has continued for more than a month, and uncertainty over when supplies will normalize is increasing anxiety in the field."Some speculate the shift in business strategy had an effect… industry eyes timing of supply normalizationThe situation has also prompted speculation that, beyond temporary production issues, changes in Daiichi Sankyo's business priorities may have contributed to the shortage.As competition in the Lixiana market is set to intensify with generic versions of Lixiana expected to enter Korea's National Health Insurance reimbursement system in the second half of the year, Daiichi Sankyo has been focusing its investments on oncology, particularly antibody-drug conjugates (ADCs).The company has been expanding its global oncology business around ‘Enhertu (trastuzumab deruxtecan)’ and ‘Datroway (datopotamab deruxtecan)’ while also accelerating the development and commercialization of follow-on ADCs. As a result, some have questioned whether production and supply of cardiovascular products approaching patent expiry have become a lower priority than in the past.However, as this supply delay is known to have occurred simultaneously in the global market rather than being limited to South Korea, the prevailing view is that it is difficult to interpret it as being directly linked to patent expirations or domestic sales strategies. There is little evidence to suggest the company intentionally reduced supplies to the Korean market because of patent expiration or domestic commercial strategy.The key question now is when supplies will return to normal. If the disruption continues, it could increase the burden of switching prescriptions for hospitals and pharmacies while also causing inconvenience for patients who have been taking these therapies over the long term.Daiichi Sankyo Korea is reportedly coordinating the timing for resuming domestic supply based on the completion of maintenance work at the manufacturing facility and product-specific shipment schedules.
- Company
- Sales of 7 Celltrion products surpass ₩100 Billion in 1H
- by Chon, Seung-Hyun Jul 28, 2026 08:57am
- Celltrion's recently launched products have become a major growth engine. In the first half of this year, sales from newer products exceeded those of legacy products by more than 60%. 8 of the company's 9 newer products recorded year-on-year sales growth, more than compensating for the stagnation of older products and driving overall revenue growth. The strong performance of higher-margin products also significantly improved profitability.According to Celltrion on July 27, seven of its 12 marketed products generated more than KRW 100 billion in sales during the first half of the year.The products are: Remsima, Truxima, Remsima SC, Yuflyma, Omlyclo, Stoboclo/Osenvelt, and Vegzelma.Celltrion has obtained regulatory approvals in Europe and the US for Remsima, Herzuma, Truxima, Remsima SC, Zymfentra, Yuflyma, Vegzelma, Steqeyma, Stoboclo/Osenvelt, Omlyclo, Avtozma, and Eydenzelt.Remsima is a biosimilar referencing the autoimmune disease therapy Remicade. Herzuma and Truxima are biosimilars of the oncology drugs Herceptin and MabThera, respectively. Remsima SC is a subcutaneous (SC) formulation developed by Celltrion by converting the original intravenous (IV) formulation of Remsima. In the US, Remsima SC received new drug approval under the brand name Zymfentra.Yuflyma is a biosimilar version of the autoimmune therapy Humira. Vegzelma and Steqeyma reference the original drugs Avastin and Stelara, respectively. Stoboclo and Osenvelt are biosimilars of the bone disease therapies Prolia and Xgeva. Omlyclo is a biosimilar version of the allergy and asthma treatment Xolair, while Avtozma and Eydenzelt reference the original products Actemra and Eylea, respectively.AI-generated imageThe growth momentum of newer products significantly outpaced that of the legacy portfolio.Celltrion classifies Remsima, Herzuma, and Truxima, which were launched during the early stages of its biosimilar business, as legacy products. Biologics introduced since 2020—including Remsima SC, Yuflyma, Vegzelma, Steqeyma, Stoboclo/Osenvelt, Omlyclo, Avtozma, and Eydenzelt—are categorized as newer products.Sales from the newer portfolio rose 72.0% year over year to KRW 1.4061 trillion in the first half, up from KRW 817.3 billion a year earlier. In contrast, combined sales of the 3 legacy products edged down 0.4% to KRW 832.0 billion from KRW 835.3 billion. Sales of newer products exceeded those of legacy products by 69.0%, underscoring their role as the primary driver of the company's growth.Among the three legacy products, only Truxima posted sales growth during the first half. Truxima generated KRW 288.8 billion in sales, up 24.8% year over year. Meanwhile, sales of Remsima and Herzuma declined 6.4% and 27.3%, respectively.8 of the 9 newer products recorded higher sales in the first half of this year than last year.Remsima SC generated KRW 356.7 billion in first-half sales, up 20.3% year over year. Yuflyma’s sales increased 21.4% to KRW 309.5 billion from KRW 254.9 billion generated last year.Omlyclo and Stoboclo/Osenvelt reported eightfold and fifteenfold increases in sales, respectively, surpassing KRW 100 billion in half-year sales for the first time. Although Vegzelma's first-half sales declined 17.5% year over year to KRW 114.7 billion, it also exceeded the KRW 100 billion mark.Newer products accounted for 65% of Celltrion's sales in the second quarter of this year, up from 53% in the same period last year.The expanding contribution of newer products is directly enhancing the company’s profitability. Most of Celltrion's recently launched products are sold through the company's own direct sales infrastructure, a structure that delivers higher margins than sales conducted through overseas commercial partners.In the first half, Celltrion posted operating profit of KRW 773.7 billion, up 97.4% year over year, while revenue increased 40.8% to KRW 2.5387 trillion. Its operating margin improved to 30.5% in H1 from 21.7% a year earlier, representing an increase of 8.8 percentage points year over year.

- Company
- Calls for reform in NHI access for third-line mCRC treatment
- by Son, Hyung Min Jul 27, 2026 08:43am
- There is a growing demand in South Korea to improve access to third-line treatments under the National Health Insurance (NHI) for metastatic colorectal cancer (mCRC) to enhance patient survival.While health insurance coverage is relatively comprehensive for first- and second-line treatments in South Korea, global standard-of-care agents used in subsequent lines remain non-reimbursed. In response, health authorities have acknowledged the high unmet medical need in third-line treatment, stating that they are evaluating measures to improve patient access.On the 24th, a symposium titled "Policy Forum for Improving the Treatment Environment for Metastatic Colorectal Cancer Where Early Treatment Access Determines Survival" was held at the National Assembly, hosted by Representative Mihwa Seo of the Democratic Party of Korea.Rep. Seo stated, "In metastatic colorectal cancer, treatment timing and therapeutic access exert a direct impact on overall survival, yet reimbursed options in third-line are not available in South Korea," and added, "A rational reimbursement framework must be established to prevent patients from forfeiting treatment due to financial toxicity."A policy symposium on improving the treatment environment for metastatic colorectal cancer was held on July 24 at the National Assembly Members' Office Building.Third-line treatments are not reimbursed...Leading to drops in treatment ratesPresenting at the forum, Professor Myung Ah Lee of the Division of Oncology at Seoul St. Mary's Hospital diagnosed that as patients with metastatic colorectal cancer progress through sequential lines of therapy, acquired resistance and disease progression progressively deteriorate their systemic performance status. Dr. Lee explained that non-reimbursed drug costs starting at the third-line setting lead to a sharp decline in the proportion of patients maintaining ongoing treatment.Professor Lee pointed out, "Reimbursement is well-integrated through first- and second-line systemic chemotherapy, resulting in a manageable financial burden for patients. However, from the third-line setting onward, patients with good performance status are frequently unable to receive treatment simply because no reimbursed agents exist," and added, "An increasing number of patients are discontinuing treatment despite viable therapeutic opportunities due strictly to financial constraints."Currently, first-line therapy for metastatic colorectal cancer primarily utilizes oxaliplatin- or irinotecan-based cytotoxic chemotherapy in combination with targeted biologics. Second-line treatment involves switching to the alternative chemotherapy backbone not administered in the first-line setting.However, the treatment landscape shifts dramatically following failure of both lines. While a small subset of patients with specific biomarkers can access immune checkpoint inhibitors or targeted agents, the eligible patient population remains narrow.For the majority of patients, global standard-of-care regimens recommended in guidelines, such as the combination of 'Lonsurf (trifluridine/tipiracil)' plus 'Avastin (bevacizumab)', 'Stivarga (regorafenib)', and 'Fruzaqla (fruquintinib)', are all non-reimbursed in South Korea.Professor Lee emphasized, "In the U.S. and Europe, Stivarga, Lonsurf-Avastin combination, and Fruzaqla are recommended from the third-line setting, but none of these regimens are reimbursed in South Korea," and added, "We need a regulatory environment where therapeutics capable of preserving quality of life alongside overall survival can enter the national health insurance benefit umbrella more rapidly."Access to Next-Generation Sequencing (NGS) testing for precision oncology was also pointed out to be resolved, as the absence of genomic profiling data restricts patient eligibility for biomarker-driven novel drugs and clinical trial enrollment.Professor Lee stated, "While international practice is moving toward routine genomic profiling for patients with metastatic or recurrent solid tumors, domestic reimbursement in Korea remains restricted outside of specific cancer types," and expressed concerns that "Consequently, patients who wish to participate in clinical trials for novel therapies often cannot enroll due to a lack of genetic sequencing results."Experts urged that even in third-line and subsequent settings, clinicians must have the therapeutic flexibility to decide the sequence of care based on patient performance status, prior treatment history, and specific toxicity profiles.Professor Dong-Hoe Koo of the Division of Hematology-Oncology at Kangbuk Samsung Hospital explained, "Each therapeutic option possesses distinct efficacy as well as unique toxicity profiles, such as fatigue, thrombocytopenia, hand-foot syndrome, and hypertension,” and added, “Drug selection should be personalized according to prior treatment exposure and individual patient vulnerability to specific adverse events."Dr. Koo added, "Because third-line therapeutics established as global standards remain non-reimbursed in Korea, a patient's financial status directly dictates therapeutic choices," and added, "Reimbursement access must be improved so that patients with preserved performance status can gain survival opportunities through third-line and subsequent therapies."(From left) Min-Jung Kim, Administrative officer at the Ministry of Health and Welfare; So-Young Lee, Manager of the Pharmaceutical Benefit Management Division at HIRA; Professor Dong-Hoe Koo of Kangbuk Samsung Hospital; and Professor Myung Ah Lee of Seoul St. Mary's Hospital.Discussions continue on regulatory reforms to address unmet medical needDuring the panel discussion, structural limitations in the pharmacoeconomic evaluation process and directions for regulatory reform emerged as key agenda items.Reporter Yun-Ho Eo of DailyPharm pointed out that novel therapeutics approved via placebo-controlled clinical trials face structural disadvantages during pharmacoeconomic evaluations, as they are forced to compete against outdated comparator drugs.Eo stated, "Recently, we have observed significant delays between passing the Cancer Disease Review Committee (CDRC) and being tabled before the Pharmaceutical Reimbursement Evaluation Committee (PREC)," and added, "Special policy mechanisms need to be considered for diseases where a new treatment landscape has formed, but cost-effectiveness is inherently difficult to prove due to outdated comparator drugs."Eo added, "Even if a drug does not qualify for a full pharmacoeconomic evaluation waiver or an elevated Incremental Cost-Effectiveness Ratio (ICER) threshold, we need flexible regulatory pathways for drugs occupying an intermediate tier," and added, "Multinational pharmaceutical subsidiaries in Korea must also actively negotiate with their global headquarters rather than abandoning reimbursement due to challenging external environments."Government representatives acknowledged the severity of the coverage gap in third-line metastatic colorectal cancer. They explained that regulatory reforms are underway to incorporate high unmet medical needs into reimbursement decision-making.Lee stated, "We are aware of the reality that patients face due to a lack of options in third-line therapy, and we feel a deep sense of responsibility," and added, "Under the principle that unmet medical needs in life-threatening severe diseases should be evaluated through dedicated mechanisms, we are accelerating regulatory reforms."The government currently operates a "conditional early listing with post-evaluation" pathway for high-cost novel drugs that demonstrate substantial unmet medical need despite limited clinical evidence. Separately, health authorities are evaluating flexible ICER thresholds for therapies indicated for severe diseases where applying standard ICER benchmarks is unfeasible.Lee stated, "A research initiative evaluating the application of flexible ICER thresholds is scheduled for completion around November of this year, after which implementation will proceed," and added, "Even before the completion of this research, working-level staff is thoroughly reviewing data so that the evaluation committee can adequately consider disease characteristics and unmet needs."Lee noted, "In reviewing third-line treatments, including Fruzaqla, we are re-examining the reasons why previous agents failed to secure reimbursement, the criteria applied at the time, and our newly evolved administrative procedures," and added, "Even before the research findings are published, we will fully consider the third-line colorectal cancer reimbursement gap within the committee's existing criteria and authority."Min-Jung Kim, Manager of the Division of Health Insurance Benefits at the Ministry of Health and Welfare, noted, "Because National Health Insurance operates within a finite budget, we must strike a balance between patient access and fiscal sustainability," and added, "We are striving to establish rational reimbursement solutions by comprehensively reviewing the clinical value of therapeutics and their impact on patient quality of life."Kim added, "It is important for pharmaceutical companies to demonstrate proactive negotiation and a willingness to improve patient access," and concluded, "The government will continue driving regulatory improvements to ensure that essential therapeutics are supplied to patients more rapidly."
- Company
- Voxzogo may be prescribed at tertiary hospitals in Korea
- by Eo, Yun-Ho Jul 27, 2026 08:43am
- Voxzogo, Korea’s first treatment for children with achondroplasia, has gained a foothold in general hospitals in Korea.According to industry sources, Voxzogo (Vosoritide), which Samoh Pharm licensed from BioMarin Pharmaceutical, has been approved by the drug committees (DCs) at 10 hospitals nationwide, including four of Korea's ‘Big 5’ hospitals—Samsung Medical Center, Seoul National University Hospital, Asan Medical Center, and Severance Hospital—as well as Keimyung University Dongsan Medical Center and Chungnam National University Hospital.Following its inclusion on the National Health Insurance reimbursement list last month, the therapy has been rapidly expanding its prescribing footprint.Voxzogo targets the FGFR3 signaling pathway, which is involved in the underlying cause of achondroplasia. In Korea, it was designated as the 10th product under the Ministry of Food and Drug Safety's Global Innovative Products on Fast Track (GIFT) program and approved for pediatric patients aged 4 months or older with achondroplasia with open growth plates.Under the reimbursement criteria that took effect in June, eligible patients are children aged 4 months or older with a confirmed FGFR3 mutation by genetic testing. Patients must have open growth plates and must not have undergone limb-lengthening surgery.To continue treatment, patients are evaluated every 6 months. Therapy is discontinued if the growth plates close or if the patient's annual growth velocity fails to meet the required threshold.Achondroplasia is not simply a condition characterized by short stature. In addition to impaired growth, patients may experience disproportionate body proportions, orthopedic complications, neurological complications, respiratory problems, limitations in daily functioning, and psychosocial burdens.Accordingly, the goal of treatment extends beyond increasing height. The therapy holds significance in that it improves growth while lowering the burden of long-term health management, thereby improving the quality of life for patients and caregivers.Meanwhile, Voxzogo demonstrated its efficacy in a Phase III clinical trial. In a study involving 121 pediatric patients with achondroplasia aged 5 to 14.9 years, the Voxzogo group showed an increase in annualized growth velocity of 1.40 cm/year from baseline after 52 weeks of treatment, whereas the placebo group experienced a decrease of 0.17 cm/year, demonstrating a statistically significant improvement of 1.57 cm/year.
- Company
- Invossa still mired in legal battles 7 years after license revocation
- by Kim, Jin-Gu Jul 24, 2026 08:21am
- Kolon TissueGene's knee osteoarthritis candidate TG-C failed to demonstrate improvements in pain and physical function in its U.S. Phase III trial. The clinical setback is expected to have a significant impact on the ongoing litigation involving Invossa Invossa-K Inj (Invossa), which is being pursued against Kolon TissueGene and Kolon Life Science. The two companies are currently involved in a total of 55 Invossa-related lawsuits with claims exceeding KRW 100 billion.Kolon TissueGene and Kolon Life Science in 55 damages lawsuits worth KRW 104.1 billionAccording to Korea's Financial Supervisory Service on July 23, Kolon TissueGene and Kolon Life Science are currently involved in 55 lawsuits related to Invossa—32 involving Kolon Life Science and 23 involving Kolon TissueGene. Most are claims seeking damages related to Invossa, with the total amount in dispute near KRW 104.1 billion.The plaintiffs who filed the lawsuit claim that they suffered physical and financial damages due to the change in Invossa's main cell line, which was revealed in 2019.Invossa drew global attention after receiving approval from Korea's Ministry of Food and Drug Safety (MFDS) in July 2017 as the world's first gene therapy for knee osteoarthritis. TG-C is the product's US development name.However, during the US Phase III trial in March 2019, it was discovered that one of the product's principal components, originally described as cartilage-derived cells, had in fact been replaced with kidney-derived cells that carry tumorigenic potential. The MFDS revoked Invossa's marketing authorization in April 2019, while the US Food and Drug Administration (FDA) placed the clinical trial on hold. The FDA later lifted the clinical hold in April 2020.Kolon TissueGene is currently facing 16 shareholder lawsuits seeking damages for investment losses. A total of 2,048 shareholders are participating in these cases, with claims amounting to KRW 55.5 billion. In addition, 931 patients who received Invossa have filed 6 damages lawsuits against Kolon TissueGene for KRW 12.2 billion.Kolon Life Science is also engaged in large-scale litigation with both shareholders and patients. Shareholders have filed 21 lawsuits seeking KRW 24.1 billion in damages, while 941 patients have filed 10 lawsuits seeking KRW 12.4 billion. The company is also being sued by multiple domestic insurers seeking reimbursement through subrogation claims.Industry and legal experts expect the latest US Phase III results to have little direct impact on the outcome of the damages lawsuits. However, they say the failed trial could increase the companies' financial and legal burdens when courts determine damages or during settlement negotiations.Most cases still await first-instance rulings at 7 years…Kolon loses first patient lawsuitAlthough more than 7 years have passed since the first wave of litigation began, the vast majority of cases have yet to receive even a first-instance ruling.A recent damages lawsuit filed by Invossa patients, however, resulted in the first trial court decision. On July 9, the Seoul Central District Court ruled entirely in favor of 139 patients who sued Kolon TissueGene and Kolon Life Science for damages.The court found that Invossa had been manufactured using kidney-derived cells rather than the cartilage-derived cells identified in the original marketing application, recognizing this as a “manufacturing defect.” It also concluded that marketing and selling the product while labeling it as containing cartilage-derived cells violated both the Pharmaceutical Affairs Act and the Act on Fair Labeling and Advertising. Accordingly, the court held the companies liable for both economic damages and emotional distress suffered by the patients. It rejected Kolon's argument that the defect could not have been identified based on the scientific knowledge available at the time of manufacture.Despite prevailing at trial, it will take more time for patients to receive compensation, as Kolon has appealed the ruling to the Seoul High Court. In addition, Kolon has blocked enforcement actions such as the seizure and collection of claims by patients by filing for a stay of execution.Most shareholder lawsuits likewise remain at the first instance stage. Three lawsuits against Kolon TissueGene have already concluded after plaintiffs withdrew their claims or courts recommended settlement. In 4 of the company's 16 shareholder lawsuits, however, trial courts ruled in favor of Kolon. The plaintiffs have appealed, with those cases now before the appellate court.By contrast, the dispute with Mitsubishi Tanabe Pharma over the return of upfront payments and damages arising from the terminated licensing agreement was resolved early on. In 2016, Kolon Life Science signed a technology licensing agreement with Mitsubishi Tanabe worth up to JPY 50 billion and received an upfront payment of JPY 2.5 billion. Mitsubishi Tanabe terminated the agreement in December 2017 and filed arbitration with the International Chamber of Commerce (ICC) the following April. After the changed cell line issue emerged in May 2019, it was added as an additional ground for termination. Ultimately, Kolon returned the JPY 2.5 billion upfront payment along with JPY 134 million in damages in April 2021.Administrative appeal over ‘Invossa license revocation’ still pending before Supreme CourtKolon Life Science is also pursuing administrative litigation in addition to the civil lawsuits. After the Ministry of Food and Drug Safety (MFDS) revoked Invossa's marketing authorization in 2019, the company filed an administrative lawsuit challenging the decision.Both the Seoul Administrative Court (first instance) in February 2021 and the Seoul High Court (second instance) in February 2024 ruled in favor of the MFDS. The courts found that the discrepancy between the cell component described in the marketing authorization application and the one actually detected constituted a material defect, dismissing Kolon's claims. Kolon subsequently appealed to the Supreme Court, where the case remains pending.In addition to the lawsuit over the revocation of Invossa's marketing authorization, Kolon Life Science also filed lawsuits seeking to ▲overturn the MFDS Commissioner's revocation of its clinical trial authorization, ▲ invalidate the Daejeon Regional Office of Food and Drug Safety's order to recall and dispose of Invossa, and ▲cancel the Ministry of Health and Welfare's and the Ministry of Science and ICT's orders to recover government research funding.Among these, the lawsuits challenging the revocation of the clinical trial authorization and the recall and disposal order were voluntarily withdrawn by Kolon Life Science. Meanwhile, the lawsuit seeking to overturn the government's recovery of research funding ultimately ended in Kolon's favor after reaching the Supreme Court. The company had received KRW 1.25 billion in government funding during the development of Invossa.Executives including former Chairman Woong-yeol Lee and CEO Woosok Lee were acquitted in criminal casesCriminal proceedings against Honorary Chairman Woong-yeol Lee, former CEO Lee Woo-seok, and other Kolon Life Science executives concluded with acquittals.Honorary Chairman Woong-yeol Lee and former CEO Lee Woo-seok were indicted on charges including violations of the Capital Markets Act and the Pharmaceutical Affairs Act. Prosecutors alleged that they concealed the US Food and Drug Administration's clinical hold order, attracted investment through the company's listing, manipulated the share price, and made false disclosures in violation of capital markets regulations.Both the first- and second-instance courts acquitted the defendants. The trial court found that the evidence presented was insufficient to conclude that the defendants knowingly concealed the cell line change or intentionally made false disclosures to attract investment.The appellate court (second instance) ruling was also handed down in February of this year. The Seoul High Court also upheld the acquittals. The appellate court ruled that the misunderstanding regarding the origin of the cells was recognized only after the product had already been manufactured and marketed, characterizing it as an error made during the development process rather than a deliberate cover-up. The acquittals became final after prosecutors decided not to appeal to the Supreme Court.The former head of Kolon Life Science's Bio New Drug Research Center and its former medical team leader were also acquitted by the Supreme Court. They had been charged with obstruction of official duties by fraudulent means, fraud under the Act on the Aggravated Punishment of Specific Economic Crimes, and violations of the Subsidies Management Act.The trial court ruled that there was insufficient evidence to conclude that they had interfered with the MFDS's review process or fraudulently obtained government R&D subsidies by deceiving government evaluators. However, the former medical team leader was found guilty of providing entertainment to an MFDS official and was fined KRW 10 million. The appellate court likewise found no evidence of intentional submission of false data or subsidy fraud, and the Supreme Court upheld those findings by dismissing the prosecution's appeal. The bribery conviction, however, was ultimately upheld, leaving the fine in place.
- Company
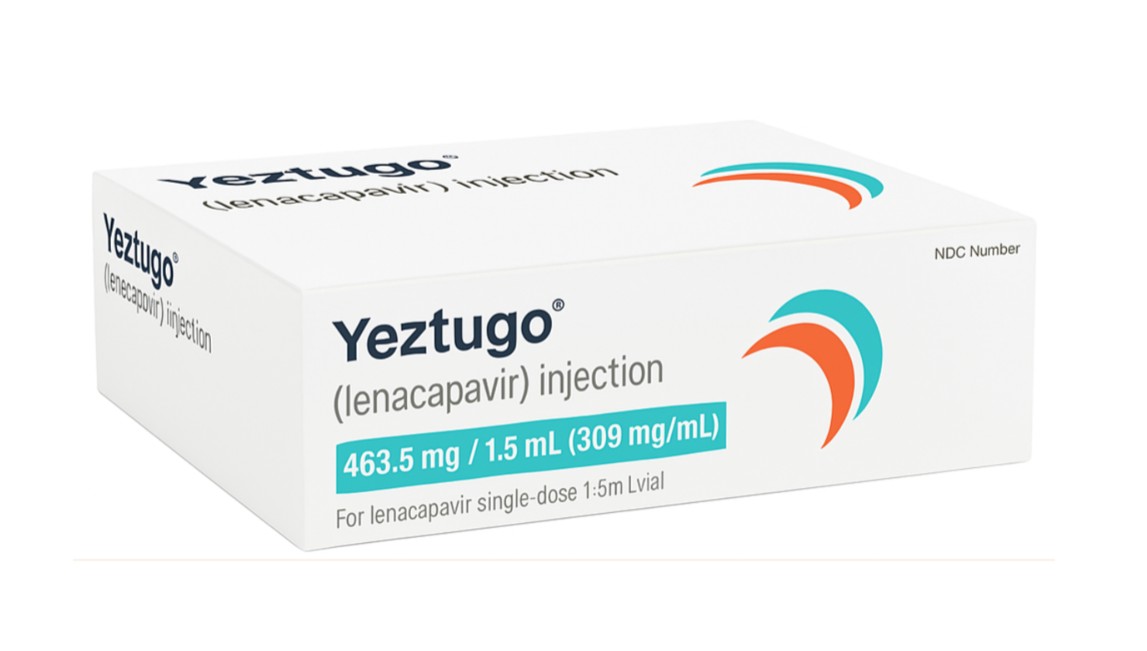
- HIV treatment evolving beyond 'once-daily'
- by Son, Hyung Min Jul 23, 2026 09:09am
- Competition in human immunodeficiency virus (HIV) treatment development is shifting from viral suppression efficacy toward enhancing patient dosing convenience.As once-daily oral therapy has become the standard of care, clinical outcomes for once-weekly oral treatments and twice-yearly preventive injections are being disclosed, demonstrating shifts in both therapeutic and preventive strategies.According to industry sources on July 23, major global pharmaceutical companies are scheduled to present research results on long-acting treatment strategies, including once-weekly HIV therapies and twice-yearly pre-exposure prophylaxis (PrEP), at the 26th International AIDS Conference (AIDS 2026) starting on July 26 in Rio de Janeiro, Brazil.First Phase 3 success for a once-weekly oral treatmentThe most highlighted result is the Phase 3 clinical trial results of an oral combination therapy combining Gilead's lenacapavir and MSD's islatravir.Lenacapavir is a long-acting agent that inhibits viral replication by targeting the HIV capsid. Islatravir is a nucleoside reverse transcriptase translocation inhibitor (NRTTI) with a mechanism distinct from conventional reverse transcriptase inhibitors, acting to block viral replication.Gilead is expanding its long-acting strategy in both treatment and prevention based on lenacapavir. Following the launch of 'Sunlenca' as a therapeutic agent, the company applied a separate product name, 'Yeztugo,' for its preventive indication.Until now, long-acting HIV treatment has centered on injectable agents administered up to once every two months or once-daily oral regimens. To achieve injectable-level dosing convenience with an oral formulation, both companies have been developing a once-weekly oral therapy combining the long-acting agent lenacapavir with the novel-mechanism islatravir.The global Phase 3 trials, designated ISLEND-1 and ISLEND-2, evaluated the efficacy and safety of switching from conventional daily oral therapy to once-weekly treatment (lenacapavir + islatravir) in virologically suppressed people living with HIV. Both studies demonstrated non-inferiority in viral suppression efficacy at 48 weeks compared to existing daily oral regimens.ISLEND-1 is a study comparing maintenance therapy with Gilead's 'Biktarvy' (bictegravir + emtricitabine + tenofovir alafenamide) against switching to lenacapavir + islatravir following Biktarvy administration.Among 607 patients evaluated, none in the lenacapavir + islatravir arm failed to maintain viral suppression at week 48, compared with 1 patient (0.3%) in the Biktarvy maintenance arm.Similar results were confirmed in ISLEND-2, which enrolled patients maintaining various daily oral regimens. At week 48, the proportion of patients with HIV RNA at or above 50 copies/mL (the threshold for viral suppression) was 1 patient (0.3%) in the lenacapavir + islatravir group and 4 patients (1.3%) in the baseline regimen group.Safety profiles were also comparable to existing therapies. Concerns regarding decreases in lymphocyte and CD4+ T-cell counts, previously raised during high-dose development, were not observed in these trials.If approved by regulatory authorities, lenacapavir + islatravir will become the world's first once-weekly oral HIV treatment.Twice-yearly era for prevention… Expanding long-acting strategiesGilead 'Yeztugo'In addition to HIV treatment, long-acting strategies are expanding into pre-exposure prophylaxis (PrEP) to prevent infection.Gilead will also present long-term follow-up data for its twice-yearly HIV prevention injection, Yeztugo. This study evaluated post-approval long-term preventive efficacy and real-world persistence, focusing on confirming the clinical value of long-acting PrEP.n the PURPOSE 1 trial, 95% of eligible participants chose to continue with Yeztugo in the open-label extension, with zero incident HIV cases reported during the 52-week follow-up period.In PURPOSE 2 as well, 95% of participants selected Yeztugo administration, maintaining adherence rates above 90%.The twice-yearly administration regimen is recognized as a strategy that reduces pill burden and improves persistence compared to daily oral PrEP.Beyond efficacy into a 'dosing interval' competitionGSK 'Cabenuva'In addition to Gilead, GSK's injectable 'Cabenuva' (cabotegravir + rilpivirine), administered once monthly or once every two months, holds an established position in the long-acting HIV treatment market.Gilead is also pursuing the development of next-generation long-acting therapeutics, including once-weekly oral treatments, twice-yearly preventive injections, and combinations of lenacapavir with broadly neutralizing antibodies (bNAbs).MSD also recently secured FDA approval for its once-daily islatravir-based combination tablet, 'Idvynso,' while concurrently advancing the development of a once-monthly oral PrEP candidate (MK-8527) and a once-weekly oral treatment.Currently, daily oral medication is the standard of care in HIV treatment. As development progresses from long-acting injectables to once-weekly oral therapeutics, options personalized to patients' lifestyles and treatment preferences are expected to become increasingly diverse.