- LOGIN
- MemberShip
- 2026-06-13 20:43:19

- Policy
- Incrementally modified drugs unaffected by price reform
- by Jung, Heung-Jun May 06, 2026 03:28pm
- Under the government’s drug pricing system reform, the insurance pricing premium rates for incrementally modified drugs and their combination products are expected to remain unchanged. Only some conditions related to the duration of the premium are likely to be adjusted.As the pricing calculation rate for generics is set to drop to 45%, domestic companies are expected to show greater interest in developing incrementally modified drugs.According to industry sources on the 6th, during working-level discussions between the government and the pharmaceutical industry, a consensus was reached not to lower the premium for incrementally modified new drugs.Although maintaining the current premium was discussed at the Health Insurance Policy Deliberation Committee (HIPDC) in November last year, the point was excluded from the reform plan approved by the HIPDC in March this year.This led to concerns within the industry that the premium might also be reduced along with the lower pricing calculation rate. There were concerns that lowering the premium could eliminate incentives for R&D investment.Under the current pricing system, incrementally modified drugs receive a premium on a base pricing rate of 53.55%, resulting in a final price of 70%. For new dosage or administration forms, a 58.9% premium is applied, resulting in a price of 77%.For incrementally modified combination drugs, pricing is calculated as the sum of pre-patent-expiry prices of each component. Innovative pharmaceutical companies receive 68% of that sum, while general pharmaceutical companies receive 59.5%.The government is not expected to significantly adjust these premium rates. Instead, it is reported that the preferential premium rate for combination drugs by general pharmaceutical companies—currently a 59.5% sum—will be slightly adjusted to a 60% sum.As the generic pricing rate is reduced from 53.55% to 45% while the incrementally modified drug premium remains unchanged, the price gap between generics and modified drugs is expected to widen further.The conditions for the premium duration are expected to be simplified. Previously, a one-year premium could be extended in two-year increments through conditional approvals and reviews.Going forward, a basic one-year premium will be granted, with an additional three-year extension if domestic manufacturing criteria are met. If no follow-on generics are listed thereafter, the premium may continue.Modified drugs and their combination products that are domestically produced and face no market competition will be able to maintain their premium drug prices for a long time.
- Policy
- What's the reason behind domestically developed CAR-T 'Rimqarto' obtaining Phase 3 waiver?
- by Lee, Tak-Sun May 04, 2026 10:33am
- CAR-T therapy Rimqarto (source: Curocell)Rimqarto (anbalcabtagene autoleucel, Curocell), the first domestically developed CAR-T therapy to be approved in South Korea, has been granted a waiver for Phase 3 clinical trials.This decision is interpreted as the result of a comprehensive consideration of the unique characteristics of the drug as a third-line treatment for lymphoma, as well as the ethical dilemmas associated with comparative clinical trials against existing therapies.According to the results of the Ministry of Food and Drug Safety (MFDS)'s Central Pharmaceutical Affairs Advisory Committee (CPAC) meeting held on April 2, it was concluded that it is appropriate to waive the Phase 3 clinical trial for the new CAR-T (Chimeric Antigen Receptor T-cell) therapy 'Rimqarto' and replace it with post-marketing surveillance.CPAC members highlighted that while Rimqarto has the same basic mechanism as existing CAR-T agents, it introduces a novel mechanism that simultaneously inhibits PD-1 and TIGIT to prevent T-cell exhaustion.According to a recently disclosed meeting report, one member highly evaluated the product's efficacy, stating, "The response rates were better than the clinical results of previously approved therapies, particularly with a high proportion of patients achieving complete remission (CR) and encouraging long-term survival results."Regarding safety, no specific issues were found besides the adverse events typically reported in similar agents (such as CRS and ICANS), and deaths during the trial were judged to have a low correlation with the drug.On the highly debated issue of giving a 'conditional pass for Phase 3 clinical trials,' the committee reached a consensus that it is "practically impossible." First, they viewed it as lacking ethical validity. Given that already-proven CAR-T products are approved and in use, administering a less effective control drug to patients was deemed unethical.The difficulty of patient recruitment was also considered. The patient population in the third-line lymphoma treatment phase has a low survival rate and a small number of candidates, making it extremely difficult to conduct large-scale confirmatory trials that include a control group. Consequently, the CPAC concluded that it is more rational to continuously verify safety and efficacy using Real-World Data (RWD) collected in clinical settings or Post-Marketing Surveillance (PMS), rather than mandating a Phase 3 trial.Based on this CPAC advisory, the MFDS finalized the approval conditions for Rimqarto. The committee agreed that the "product approval is appropriate, given that it is a third-line lymphoma treatment," and decided to disclose the meeting report anonymously.This decision served as a stepping stone toward the rapid supply of an independently developed domestic CAR-T therapy to the field. It is expected to provide new treatment opportunities for patients with severe hematologic cancers who do not respond to existing treatments.Meanwhile, 'Rimqarto Inj' is an orphan drug for the treatment of adult patients with diffuse large B-cell lymphoma (DLBCL) and primary mediastinal B-cell lymphoma (PMBCL) that is relapsed or refractory after two or more systemic therapies.Rimqarto works by inserting genetic information into the patient's immune cells (T-cells) to enable them to recognize CD19, a surface antigen on B-cells, and then re-injecting these cells into the patient's body to identify and destroy cancer cells expressing CD19. It is designed to inhibit the expression of immune checkpoint receptors PD-1 and TIGIT, thereby blocking cancer cells' immune evasion and inducing enhanced, sustained T-cell responses to increase anti-tumor effects.
- Policy
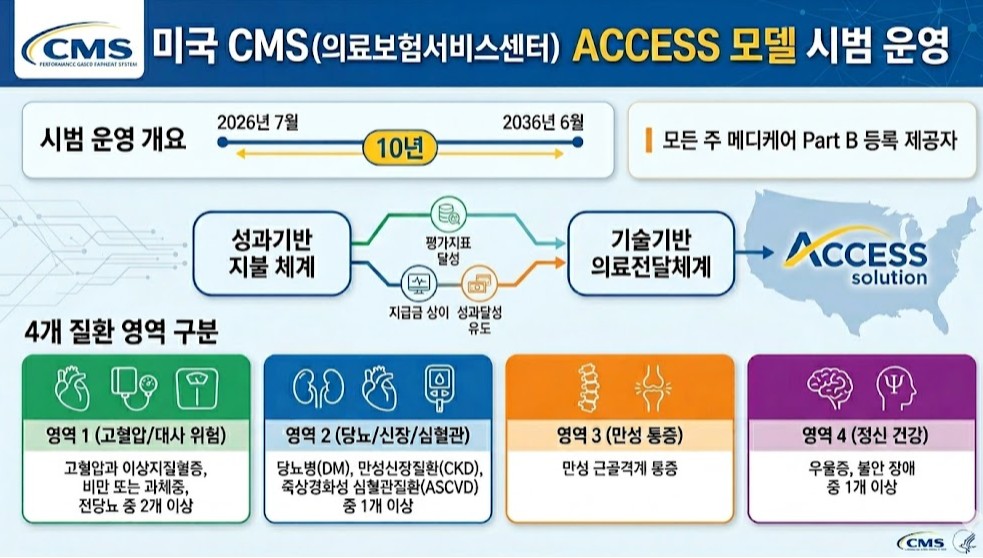
- US tests pay-for-performance to address fee-for-service limits
- by Jung, Heung-Jun May 04, 2026 10:33am
- To address the issues of an aging population and rising healthcare costs, the U.S. will launch a pilot program for a pay-for-performance system nationwide starting this July.This model links payments to the achievement of chronic disease management metrics and represents an attempt to overcome the limitations of the traditional fee-for-service (FFS) system.On the 30th, Soo-min Kwon, a senior researcher at the Health Insurance Review and Assessment Service (HIRA) Benefits Policy Research Division, published the policy implications of “The US’s New Performance-Based Payment Model for Chronic Disease Management” through an HIRA Issue report.The US Centers for Medicare & Medicaid Services (CMS) will pilot the ACCESS (Advancing Chronic Care with Effective, Scalable Solutions) model from July this year through June 2036. This model combines performance-based payment systems with technology-enabled healthcare delivery.All Medicare Part B providers across all states are eligible. Patients are categorized into four groups;▲ those with at least two of hypertension, dyslipidemia, obesity/overweight, or prediabetes, ▲ those with at least one of diabetes (DM), chronic kidney disease (CKD), or atherosclerotic cardiovascular disease (ASCVD); ▲ chronic musculoskeletal pain; ▲ those with at least one of depression or anxiety disordersParticipating institutions provide integrated services including clinical consultations, lifestyle interventions, education, and medication management. They also conduct clinical improvement activities, collecting outcome data through remote monitoring tools and implementing interventions using digital health platforms.Final control and minimum improvement targets are set for each evaluation metric, with targets adjusted annually over the treatment period.Institutions receive payments for managing patients over a one-year period. During the initial six months of treatment, they receive 50% of the annual payment in monthly installments, while the remaining 50% is paid based on whether performance targets were met and whether the patient received services for the same condition from other healthcare providers.The payment structure differs between the initial treatment year and subsequent management phases to incentivize early achievement of clinical outcomes.Kwon explained, “This represents a shift from service-based to performance-based payment systems, redesigning reimbursement criteria around clinical results rather than service quantity.”She added, “In line with global trends in payment system reform, this could serve as a reference model for designing integrated chronic disease payment systems in Korea and ensuring the sustainability of the national health insurance finances.”
- Policy
- Kwangdong secures first generic exclusivity for Tagrisso
- by Lee, Tak-Sun May 04, 2026 10:33am
- Domestic pharmaceutical companies are intensifying their push into the KRW 110 billion non-small cell lung cancer (NSCLC) treatment market centered on Tagrisso (osimertinib). Following Chong Kun Dang’s first generic approval in January, Kwangdong Pharmaceutical has also joined the ranks of companies obtaining first generic exclusivity, marking the start of a full-scale race to capture the market.Kwangdong Pharmaceutical’s ‘Ktinib Tab’ Approved… Second after Chong Kun DangAccording to the Ministry of Food and Drug Safety, Kwangdong Pharmaceutical obtained first generic exclusivity on April 30 for its osimertinib mesylate product ‘Ktinib Tab (40mg, 80mg).’ This is the second such approval following Chong Kun Dang’s ‘Otinib Tab,’ which received approval on January 27.The drug’s indications include, as a monotherapy: ▲Adjuvant treatment following complete resection in patients with NSCLC harboring EGFR exon 19 deletions or exon 21 (L858R) substitution mutations ▲Treatment of patients with unresectable locally advanced (Stage III) NSCLC harboring EGFR exon 19 deletions or exon 21 (L858R) substitution mutations whose disease has not progressed during or after platinum-based chemoradiotherapy ▲First-line treatment of patients with locally advanced or metastatic NSCLC harboring an EGFR exon 19 deletion or exon 21 (L858R) substitution mutation ▲Treatment of patients with EGFR T790M-positive locally advanced or metastatic NSCLC who have previously been treated with an EGFR-TKI.Additionally, as a combination therapy, it is indicated for the first-line treatment of patients with locally advanced or metastatic non-squamous NSCLC harboring EGFR exon 19 deletions or exon 21 (L858R) substitutions, in combination with pemetrexed and platinum-based chemotherapy. These indications are identical to the original Tagrisso.Like Chong Kun Dang, Kwangdong succeeded in circumventing Tagrisso’s formulation patent (set to expire January 2035), thereby securing first generic exclusivity. As a result, both companies will hold a roughly 9-month market exclusivity period starting from the expiry of the substance patent.First, generic exclusivity is granted when a company successfully challenges a patent and meets the criteria of being among the first to file for approval. Both companies achieved patent circumvention in September last year by winning a passive scope confirmation trial regarding the formulation patent. AstraZeneca has since appealed, and a lawsuit to cancel the ruling is currently ongoing at the Intellectual Property High Court.The approval applications were also submitted simultaneously to the MFDS, meaning both companies met the requirements for exclusivity.The exclusivity period for Ktinib (during which sales of identical drugs are restricted) is set from December 28, 2033, to September 27, 2034.Since the original drug Tagrisso’s substance patent remains valid until 2033, immediate market entry is not possible. However, both companies plan to enter the market immediately upon patent expiry, block latecomers, and capture the KRW 110 billion market.Tagrisso vs. Leclaza… generics join the competitionCurrently, the domestic NSCLC treatment market is dominated by AstraZeneca’s original drug Tagrisso and Yuhan’s domestic new drug Leclaza, which are competing fiercely for market share. With Chong Kun Dang and Kwangdong preparing to launch generics, the market landscape is expected to become even more complex.A pharmaceutical industry insider analyzed, “Given the high commercial potential of the product, generic companies are continuing to challenge the patents. With Chong Kun Dang and Kwangdong securing generic exclusivity, the generic market is highly likely to evolve into an initial two-horse race starting in 2033
- Policy
- Novartis Korea's SMA therapy undergoes expedited approval review
- by Lee, Tak-Sun Apr 29, 2026 03:48pm
- Product photo of onasemnogene abeparvovecOnasemnogene abeparvovec, Novartis Korea’s new drug for spinal muscular atrophy (SMA), has been designated as an item for the Ministry of Food and Drug Safety (MFDS)’s Global Innovative Products on Fast Track (GIFT).According to industry sources on the 28th, the MFDS designated the drug as the 67th GIFT item on the 8th and initiated a full-scale expedited approval review. This designation is based on recognition of the drug's innovation in treating rare diseases and its improved efficacy compared to existing treatments. The brand name of onasemnogene abeparvovec is known asItvisma.The version of this drug approved for GIFT this time changes the administration route from the existing intravenous (IV) formulation, Zolgensma, to an intrathecal (IT) injection.Unlike the original Zolgensma, which was primarily designed for use in infants and young children due to weight restrictions (under approximately 13.5 kg), this new formulation has been submitted for all SMA patients of any age who have biallelic mutations in the Survival Motor Neuron 1 (SMN1) gene. New formulation is garnering significant anticipation because it offers the opportunity for gene therapy to pediatric patients aged 2 and older and to adult patients.Onasemnogene abeparvovec utilizes an adeno-associated virus serotype 9 (AAV9) vector to deliver a functional SMN1 gene directly to the patient's target cells. This mechanism addresses the disease's fundamental cause by inducing continuous expression of the deficient SMN protein.The MFDS decided on the GIFT designation based on the drug's outstanding motor neuron improvement effects demonstrated in clinical trials and data showing improved efficacy that complements existing treatments.The drug has already proven its value in the global market. The U.S. FDA completed its approval on November 24th of last year, and Japan's PMDA also approved on April 3rd.Following this GIFT designation, Novartis Korea will receive significant support from the MFDS throughout the approval process. Analysis suggests that the review period will be shortened by approximately 25% compared to general reviews, making domestic approval possible as early as the second half of this year or early next year.A pharmaceutical industry official stated, "The introduction of innovative new drugs into Korea is accelerating through the GIFT system. This expedited review approval is favorable news for older SMA patients who faced limitations with existing treatments."
- Policy
- Approval of new ulcerative colitis drug Velsipity near
- by Lee, Tak-Sun Apr 28, 2026 09:46am
- ‘Velsipity 2mg Tab (etrasimod),’ a new ulcerative colitis treatment being introduced in Korea by Everest Medicines Korea, cleared a key hurdle toward domestic marketing authorization.According to industry sources on the 26th, the Ministry of Food and Drug Safety recently completed the safety and efficacy review for Velsipity Tab. As the safety and efficacy review is considered the most rigorous step in the new drug approval process, the likelihood of Velsipity receiving domestic marketing authorization within the year has significantly increased.Velsipity is a next-generation selective sphingosine-1-phosphate (S1P) receptor modulator developed by Pfizer. It works by blocking the release of lymphocytes from lymph nodes, thereby preventing their migration to inflamed areas in the gastrointestinal tract.It is indicated for adult patients with moderate to severe active ulcerative colitis. In particular, its once-daily oral dosing is considered a strong competitive advantage in a treatment environment traditionally dominated by injectable therapies.Velsipity has demonstrated efficacy through global Phase III trials (ELEVATE UC 52 and ELEVATE UC 12). According to trial results, it achieved significantly higher clinical remission rates compared to placebo at both week 12 and week 52.Currently, the domestic ulcerative colitis market is dominated by biologics as well as JAK inhibitors such as ‘Rinvoq’ and ‘Xeljanz.’ If approved, Velsipity is expected to form an S1P modulator market alongside the first-in-class drug Zeposia, providing patients with a new treatment option.Everest Medicines has secured commercialization rights in the Asia-Pacific region, including Korea, through an agreement with Pfizer and has been proceeding with the domestic approval process. With the completion of the safety and efficacy review, the company is nearing the acquisition of marketing authorization.An industry official said, “Passing the safety and efficacy review means that the MFDS has recognized the clinical value of the drug. Upon final approval, Velsipity is expected to have a significant impact in the ulcerative colitis treatment field, where there is a high preference for oral medications.”

- Policy
- Innovative Pharma Company certification reform…"application opens in August"
- by Lee, Jeong-Hwan Apr 28, 2026 09:46am
- The Ministry of Health and Welfare (MOHW) plans to begin accepting applications from pharmaceutical companies as early as August to implement the reformed "Innovative Pharmaceutical Companies" certification system, following the completion of remaining administrative procedures.As both domestic and multinational pharmaceutical companies have shown strong interest in the reformed certification system, the MOHW has decided to implement an absolute evaluation method, granting the designation of an Innovative Pharmaceutical Companies to all companies that score 65 points or higher.If applications are accepted in August as scheduled, the final list of certified pharmaceutical companies is expected to be officially notified (announced) by the end of December following the review process.On the 26th, Lim Kang-seop, director of the Pharmaceutical and Bio Industry Division at the MOHW, explained the administrative plan to reform the "Innovative Pharmaceutical Companies" certification system during a meeting with KSPA News.The MOHW has issued legislative and administrative notices regarding the enforcement decree·enforcement rules for related notifications of the "Special Act on Fostering and Support of Pharmaceutical Industry" as of the 26th of last month.The Ministry plans to finish gathering opinions by the 6th of next month, followed by an internal review, a regulatory review by the Prime Minister's Office, and a review by the Ministry of Government Legislation.The reform plan will be implemented immediately upon finalization and promulgation. The MOHW plans to start accepting applications for innovative pharmaceutical companies based on the new criteria as early as the beginning of or mid-August and continue through the end of September.Key indicators for the certification review include whether the R&D-to-revenue ratio meets the standards and whether any disqualifying factors exist. Disqualifying factors include illegal drug rebates and unethical conduct by executives or employees. Exceeding the threshold in these areas will result in failure to obtain certification.Lim explained that there will be no limit on the final number of innovative pharmaceutical companies, and certification will be granted through an absolute evaluation.Lim stated, "Certification as an innovative pharmaceutical company is possible if a minimum score of 65 points is achieved. The review will be conducted as an absolute evaluation," and added, "After the Korea Health Industry Development Institute (KHIDI) conducts an initial document review of the applicant companies, those that pass the criteria will undergo deliberation by the committee. We expect to be able to notify the final list by the end of December this year."Given the high interest from both domestic and foreign pharmaceutical companies, with over 100 companies expected to apply, the specific schedule for the official notification of the list may change.Lim plans to hold briefing sessions and public-private consultations regarding the certification system for domestic and foreign pharmaceutical companies between May and June.Lim also noted that further review by the MOHW is required regarding the specific evaluation methods and score allocations for new indicators, such as supply chain stabilization for medicines experiencing supply instability.Lim stated, "This time, both quantitative and qualitative indicators will be reviewed together. Quantitative indicators are being simulated on a 5-point scale, while qualitative indicators will be evaluated according to guidelines for the reviewers," and said. "We are working with the KHIDI to determine evaluation methods suited for new indicators like supply chain stabilization."Lim further stated, "Once the detailed review guidelines are established, we plan to provide briefing sessions or further guidance. A briefing session could be held around late May or early June," and added, "There are requests to include compliance management indicators related to pharmaceutical rebates in the review process, but these are already reflected in the qualitative indicators."Lim concluded by stating that "The qualitative evaluation already accounts for instances where a pharmaceutical company has independently adopted a Compliance Program (CP) and operated a compliance management program following the discovery of rebates," and added, "The legislative notice contains only the broad framework of review items, and the method of qualitative evaluation will be determined by the KHIDI's reviewer guidelines and the items in the documents submitted by the companies. Even if there were past rebates, companies will be able to appeal by demonstrating how they have since strengthened internal controls."Meanwhile, Semi-Innovative Pharmaceutical Companies are also expected to be reviewed and selected through an absolute evaluation based on R&D standards and disqualifying factors, similar to the process for Innovative Pharmaceutical Companies.

- Policy
- Implementation of fast-track listing pilot project imminent
- by Jung, Heung-Jun Apr 28, 2026 09:46am
- The list of drugs eligible for the fast-track listing pilot project for rare disease treatments, along with contract terms, is expected to take shape in the first half of this year.Among rare disease treatments, certain drugs will be selected for initial application, and follow-up measures reflecting reimbursement after post-evaluation will be included in contract terms.According to industry sources and relevant agencies on the 27th, the National Health Insurance Service (NHIS) and the Health Insurance Review and Assessment Service (HIRA) have begun working-level preparations for implementing the fast-track listing pilot project.Fast-track listing is a government plan to reform the drug pricing system by reducing the listing period for rare disease treatments from 240 days to 100 days, with the aim of improving patient access to treatment.The Ministry of Health and Welfare announced at the end of March, through the Health Insurance Policy Deliberation Committee, that it will prioritize the pilot project this year and institutionalize it starting next year. From 2028, it will be expanded to innovative new drugs.The pilot project is expected to include selected drugs among rare disease treatments, considering factors such as patient population. NHIS and HIRA are reviewing detailed selection criteria and negotiation contract conditions.Since there are already designation criteria for orphan drugs set by the Ministry of Food and Drug Safety, additional conditions are expected to be added to these existing standards.HIRA and NHIS need to significantly simplify the existing listing process. Since it is not possible to accommodate all rare disease treatments, HIRA is carefully reviewing the criteria for selecting items.In addition, HIRA is conducting a research project through the end of the year to evaluate clinical efficacy using real-world evidence (RWE). The results will be reflected in the institutionalization of the fast-track listing system next year.Both HIRA and NHIS share the view that post-listing mechanisms must be strengthened as the listing period is significantly shortened.NHIS is focusing on strengthening follow-up measures based on post-evaluation. While reducing the drug price negotiation period with pharmaceutical companies from 60 days to one month, fast-track listing negotiations will focus mainly on expenditure caps and supply obligations.NHIS is also reviewing including clauses in negotiation contracts with pharmaceutical companies that allow for price adjustments or the re-establishment of reimbursement criteria after post-evaluations.Consequently, reaching a concrete agreement on post-listing adjustment conditions during negotiations with pharmaceutical companies is expected to be a key issue.

- Policy
- First public-private meeting on drug pricing system reform imminent
- by Jung, Heung-Jun Apr 26, 2026 01:45pm
- Photo of the Health Insurance Policy Review Committee meetingThe first meeting of the public-private consultative body, which will determine the specific details of the drug pricing system reform, is imminent. Schedules for working-group meetings are being discussed for next week, and the pharmaceutical industry is forming a Drug Pricing System Task Force (TFT).With the system implementation set for the second half of the year, strategic discussions between the government and the private sector are expected to intensify within this tight timeframe.According to industry sources on the 23rd, the Korea Pharmaceutical and Bio-Pharma Manufacturers Association (KPBMA) recently formed a Drug Pricing System Response TFT after receiving applications from all pharmaceutical member companies.Five distinct TFTs have been established, including ▲Reorganization of calculation standards ▲Essential and withdrawal-prevention-drugs ▲Raw materials ▲Innovative and semi-innovative models ▲New drugs. The public-private consultations that will decide the specifics of the drug pricing reform will be addressed primarily through these TFTs.The government is also preparing to set the negotiation table, with plans to coordinate the schedule for working-group meetings as early as next week.Among the drug pricing reform measures, the government has scheduled the implementation of price cuts for currently listed drugs, price premiums for innovative and semi-innovative models, and the management of multi-item listings for the second half of the year.This is a tight schedule if discussions are to be finalized by the end of the first half. While behind-the-scenes discussions have already begun, the process of determining details through the public-private consultative body is expected to proceed at a breathless pace.Pharmaceutical companies are particularly focused on reorganizing calculation standards, including price reductions for existing listings and multi-item management. It is reported that most pharmaceutical companies expressed interest in participating in the "calculation standards reform TFT."Among these, the most heavily debated issue is the criteria for determining the timing of price cuts for currently listed drugs. Based on the 2012 listing date, tiered price adjustments will be implemented in Stages 1 and 2; however, the interpretation of combination drugs and data-submission drugs could become a major point of argument.For instance, matters requiring negotiation include how to classify combination drugs that mix single-agent components from both Stage 1 and Stage 2, and how to categorize data-submission drugs that were listed after 2012 but share the same ingredients and administration routes as reference products listed before 2012.Furthermore, regarding 'Multi-item listing management,' where prices are significantly reduced one year after the listing of 13 or more items, it is anticipated that voices will emerge demanding exception clauses for categories such as first generics.Like previous briefings on 'International drug price comparison re-evaluation,' fierce debates over each point of items are expected during public-private consultative body meetings for drug pricing reform.
- Policy
- Will the DoctorNow Prevention Act pass the National Assembly?
- by Lee, Jeong-Hwan Apr 23, 2026 10:51am
- Whether the amendment to the Pharmaceutical Affairs Act, which prohibits non-face-to-face medical treatment platforms from operating as pharmaceutical wholesalers, can be tabled and passed during the final plenary session of the 22nd National Assembly, scheduled for the 23rd, is drawing significant attention.The bill, called the "Doctor Now Prevention Act," was originally expected to be processed in a plenary session last year alongside the amendment to the Medical Service Act to institutionalize non-face-to-face treatment. However, it has been pending for over five months due to opposition from some ruling and opposition party lawmakers, as well as the Ministry of SMEs and Startups, for the reason that it undermines the revenue-generating models of startups.On the 21st, the ruling and opposition parties agreed to holding a plenary session on the 23rd to wrap up the first half of the 22nd National Assembly ahead of the June 3 local elections.In particular, the ruling party has expressed its determination to process as many livelihood bills awaiting final disposal as possible at this closing point of the first-half session, citing that approximately 120 bills are currently pending in the plenary session.This is why all attention is on whether the inclusion of the Doctor Now Prevention Act to the Pharmaceutical Affairs Act can be tabled and passed during this plenary session.For now, the Democratic Party of Korea’s Policy Committee maintains the position that there is no reason for the bill not to be processed. Their view is that it is irrational and abnormal to delay the passage of the bill due to backlash from a specific company, especially after the bill already passed the committees, including the Health and Welfare Committee and the Legislation and Judiciary Committee, with bipartisan agreement.Furthermore, despite the strong determination of the Ministry of Health and Welfare to pass the bill, the leadership of the Democratic Party reportedly views it as unprecedented for a bill, which is only awaiting plenary processing following legitimate legislative procedures, to be suddenly modified because a related ministry, the Ministry of SMEs and Startups, raised a differing opinion, leading to inter-ministerial conflict.Nevertheless, the outlook for tabling and processing the Pharmaceutical Affairs Act amendment in the plenary session on the 23rd remains challenging. This is because the legislative opposition from lawmakers in 'Unicorn Farm,' a bipartisan research group for startups and ventures, remains intense, and internal party alignment has not been definitively settled.Consequently, the Ministry of Health and Welfare has been placed in a position where it can do nothing but wait for the National Assembly's decision. Even if a non-face-to-face treatment brokerage platform establishes and operates its own pharmaceutical wholesaler to generate revenue through management that carries a high risk of conflict of interest, the ministry has no choice but to remain a bystander.An official from the ruling party explained, "The Democratic Party’s Policy Committee views that a platform's operation of a wholesaler cannot be regarded as startup innovation or a legitimate revenue model, and thus believes it is necessary to pass the Pharmaceutical Affairs Act in its original form in the plenary session on the 23rd," and added, "This bill is also essential for the institutionalization of non-face-to-face treatment. However, the fact that some disagreements within the party remain is an issue that must be resolved promptly."