- LOGIN
- MemberShip
- 2026-05-02 11:39:34
- Policy
- New drug approval fees to increase next year
- by Lee, Hye-Kyung Dec 23, 2024 05:50am
- The Ministry of Food and Drug Safety has completed preparations to raise the fee for new drug approvals to KRW 410 million from January 1 next year. Since mid-October, the agency has formed the ‘Consultative Body for Discussions on New Drug Approval and Review Procedures' and completed the 'New Drug Approval and Review Business Procedures (Guidelines for Officials)' after 4 meetings. The MFDS expects the increase in new drug approval fees to be the first step towards improving the system to the level of the U.S. FDA and the European EMA. (From the left) Director Sang-Ae Park, Director Young-Joo Kim, Director Chun-Rae Kim, The MFDS press corp recently met with Director Young-Joo Kim, Head of the Pharmaceutical Approval Management Division at MFDS; Director Chung-Rae Kim, Head of the Pharmaceutical Policy Division at MFDS; and Director Sang-Ae Park, head of the Pharmaceutical Standardization Division at the National Institute of Food and Drug Safety Division to learn more about the 'Notification of Partial Amendment to the Fee Regulations for Approval of Pharmaceuticals, etc.' that was announced on the 8th of last month. The following is the Q&A transcript of the meeting. ▶ The MFDS issued an administrative notice of the proposed revision from September 9 to November 8, and in the process disclosed the business procedures for approving and reviewing new drug items. What progress has been made since then? “In October, we created a council to discuss the new drug approval process. A total of 32 people, including 8 from the Ministry of Food and Drug Safety, 6 from the Korea Pharmaceutical and Bio-Pharma Manufacturers Association, 9 from the Korean Research-based Pharmaceutical Industry Association, 3 from the Korea Pharmaceutical Traders Association, and 6 from the Korea Biomedicine Industry Association, took part in the council. Various discussions were made during the meeting, starting with a moratorium on fee hikes. After 4 meetings to collect opinions, the final guidelines were created. There was much industry input and I think we've reached something near of a consensus. The system is ready to be implemented.” ▶The KRPIA had strongly insisted on a moratorium on the fee hike last November, have you reached a consensus?. Director Young-Joo Kim “The MFDS is on a level where we can show the approval process, transparency, and predictability of advanced regulatory agencies such as the FDA and EMA. We have fully discussed concerns through the council, and the KRPIA has no objection. Procedurally, we have increased the number of face-to-face consultations and allow GMPs to be performed within 90 days, and I think this will be a step towards creating a system that can allow new drugs to be approved within 295 days. Some companies are already preparing for the increased fees. I think the KRW 410 million fee will be fully compensated by receiving approvals a month earlier.” ▶ You said that you had held 4 meetings, and it seems that the guidelines have changed somewhat after the meetings, such as the addition of a pre-registration process to provide supplementary materials to MFDS in advance during the first and second supplement request processes, and the allowance to request an explanatory meeting based on the submitted material. Director Young-Joo Kim “As you can see from the table of guideline changes, slight changes have been made to the guideline. The biggest difference from the draft is the addition of the preliminary consultation session before fee payment to increase the predictability of the approval process before paying the fee. However, pre-consultations are limited to one session per item. Instead, we added more face-to-face consultations, so that each supplementation to their application can be consulted and reviewed in advance. In the past, there were many claims on how the MFDS makes unexpected supplement requests at the last minute. The goal is to increase predictability in this area. Through prompt communication, we aim to shorten the approval period by reducing unnecessary waste of time. We plan to also further the approval process by formally recording the minutes of our meetings. In general, by improving the transparency and predictability of the new drug approval review process, we hope to bring new drugs to market sooner, making the industry more competitive and enabling a faster supply of medicines.” ▶ How does the prior consultation process work? Director Young-Joo Kim “In the case of new drugs, I think that companies that have already prepared enough data will proceed with the prior consultation session with the goal of applying for approval so that a dedicated team will be created from this stage. I can’t ensure the details as we haven't established the team yet, but the team will be decided based on which department should examine the safety, efficacy, and quality of each drug with consideration into the ingredients and efficacy, so we plan to proceed with a preliminary team dedicated to each item to allow the drugs to be approved. In the preliminary consultation stage, we will discuss the future schedule, the point of application submission, and the establishment of a dedicated team.” ▶ Are there any companies that have expressed their intention to apply for approval after the fee hike increase in January next year? Director Young-Joo Kim "We are aware of some companies that have applied for approval. We are receiving inquiries for preliminary consultation from the companies. I cannot disclose the names of the companies, but there are several domestic and multinational pharmaceutical companies that have expressed their intention to apply for approval.” ▶You are aiming for 295 calendar days from application to approval. Do you think this can be achieved? Director Young-Joo Kim “If there is a delay in the preparation of supplementary data, the period may exceed 295 days. This means that the term can be extended by the length of the extension period. However, we have created a lot of consultation processes to shorten the time for the companies’ supplement preparation. The goal is to shorten the time between the company preparing the supplementary data and the MFDS’s review of it as much as possible through consultation. Frequent communication with companies is expected to reduce the number of supplement requests and time consumption. It will be an opportunity to advance our system forward to a global-level review system.” ▶I understand that you are recruiting specialized reviewers for new drug approvals Director Sang-Ae Park “We have been recruiting reviewers from the 13th to the 22nd. We are hiring reviewers as soon as possible and plan to finalize the recruitment process by January. We have actively promoted our recruitment through various channels such as college of pharmacies, academic societies, and social media.” ▶ I understand that the reviewers hired this time do not include doctors. It seems that physician reviewers will also be needed. Director Chung-Rae Kim "This recruitment was made to recruit highly competent reviewers to add to our existing workforce. We opened the recruitment process to meet the need to increase the number of reviewers, and we will continue to recruit doctors and other experts periodically over the next year after completing the transition of the first- and second-level personnel. In addition, once the revision of the fee notification is finalized, we will increase the overall salary of our reviewers.” ▶ There will be some items that apply for approval in Korea for the first time, even before the FDA and EMA. Do you think the MFDS owns the capacity to approve them in 295 days? Director Chung-Rae Kim "Korea was listed on the WLA last year and is a member of ICH. I think we should believe that the competence of our reviewers stands at a global level. If there is a shortcoming of the MFDS, it is in the difficulty of recruiting human resources. Even if the new drug is first to come to Korea, I think it will be possible to approve it if the manpower is reasonably replenished along with the fee increase and the number of highly competent reviewers is filled.” Director Sang-Ae Park "I would like to emphasize that the increase in the new drug approval fee is just the start of our challenge. This is just the beginning. I don't think we can reach the FDA's review level in a short period of time, but we believe it is significant that the challenge has begun.”
- Policy
- Daewoong to replace Astellas Irribow’s gap
- by Lee, Hye-Kyung Dec 23, 2024 05:49am
- Daewoong Pharmaceutical has filled the gap for Astellas Korea’s diarrhea-type irritable bowel syndrome treatment Irribow (ramosetron hydrochloride), which was discontinued in February this year. The Ministry of Food and Drug Safety approved Daewoong Pharmaceutical's Irricol Tab 2.5㎍ (ramosetron hydrochloride) on the 20th. This follows Daewoong receiving approval for the lower dose of Irricol Tab 5㎍ on May 31, filling the gap left by Irribow’s absence. There are 23 items approved for ramosetron hydrochloride in Korea, but except for Irricol, all of them have indications only for the prevention of digestive symptoms (nausea, vomiting) caused by the administration of anticancer drugs. Until last year, Irribow was the only ramosetron hydrochloride-based formulation for the treatment of diarrhea-predominant irritable bowel syndrome. However, in November last year, Astellas announced its decision to withdraw the product from Korea for business reasons and reported the discontinuation of its supply in February of this year. In the report, Astellas indicated Daewoong Pharmaceutical's Irricol as its substitute, stating, “It is unlikely that there will be a supply shortage for the treatment of IBS as sufficient alternative preparations have been identified for the treatment of patients.” According to MFDS’s production performance data, the import earnings of Irribow 5 µg and Irribow 2.5 µg in 2023 were USD 656,666 and USD 211,020, respectively. Currently, only Daewoong Pharmaceutical is licensed to treat ramosetron hydrochloride-based irritable bowel syndrome, so it is expected to be able to secure performance in place of Astellas' Irribow. Meanwhile, other than Daewoong Pharmaceutical, Pharmbio Korea is also known to have been conducting bioequivalence trials to develop a generic version of Irribow. In April, Pharmbio Korea received approval for an open, randomized, two-arm, two-period, fasting, single-dose, oral, crossover trial in healthy adult male subjects to evaluate the bioequivalence of Pharmbio Korea’s PBK-G2401 with Astellas' RPBK-G2401. PBK-G2401 is a generic version of Irribow, which is indicated for diarrhea-predominant irritable bowel syndrome.
- Policy
- Price of Pulmicort raised again upon request
- by Lee, Tak-Sun Dec 23, 2024 05:49am
- The price of asthma and bronchitis drug ‘Pulmicort Respule Nubuliser Suspension (budesonide, AZ), which had been raised in December last year, will again be raised in just one year. The move is intended to proactively address the drug’s global market shortage. According to industry sources on the 20th, the insurance price ceiling for the drug will increase by KRW 255 from KRW 1125 to KRW 1380, effective as of January 1st, 2025. It is reported that the price adjustment was made upon the request of the ‘Private-Public Council for Addressing Unstable Supply and Demand.’ An industry insider explained, “We understand that the council proactively requested a drug price increase to secure domestic import volume as procuring the supply and demand of Pulmicort has recently been difficult in global markets including Japan and Canada.” The price increase was finalized after negotiations with the Health Insurance Review and Assessment Service’s Drug Reimbursement Evaluation Committee and the National Health Insurance Service’s price adjustment negotiations. Pulmicort's drug price was also increased in December last year, along with another same-ingredient product Pulmican. At that time, Pulmican’s price was raised from KRW 946 to KRW 1121 per vial and Pulmicort was raised from KRW 1000 to KRW 1125 to address supply and demand insecurity. Both drugs are suspension formulations that are often used to treat bronchitis, especially in infants and children, and their use increases during this season. In response, the company plans to expand the manufacturing line of the domestically manufactured Pulmican and double its production from next year. However, the drug price of Pulmicort, the imported product, has been preemptively raised to resolve the current shortage issue. It is reported that the recent shortage of Pulmicort has caused red flags in securing stocks in pharmacies nationwide.
- Policy
- Gilead starts trial for its autoimmune disease drug in KOR
- by Lee, Hye-Kyung Dec 20, 2024 05:47am
- The B- and T-Lymphocyte Attenuator (BTLA) autoimmune disease treatment will enter clinical trials in Korea. The Ministry of Food and Drug Safety approved Gilead Sciences' application for a ‘multicenter, randomized, placebo-controlled Phase 1b clinical trial to evaluate the safety, tolerability, pharmacokinetics, immunogenicity, and pharmacodynamics of multiple ascending doses of GS-0272 in adult participants with rheumatoid arthritis.’ The trial will be conducted at Chungnam National University Hospital, Seoul National University Hospital, and Ajou University Hospital. Gilead has been conducting a Phase I trial on GS-0272 since September 2023 in the United States, Georgia, Moldova, and the United Kingdom. The global phase I trial will run until June next year, and Korea will participate for 6 months from this month. The global Phase 1 will enroll 87 patients worldwide, including 8 in Korea. The Phase 1 study of GS-0272, which evaluates the safety and tolerability of multiple doses in patients with rheumatoid arthritis, aims to evaluate the safety and tolerability of multiple ascending doses of GS-0272 in patients with rheumatoid arthritis and to characterize the pharmacokinetics of GS-0272 after multiple doses. BTLA agonists are drugs that treat autoimmune diseases caused by an overreaction of immune cells by enhancing BTLA's ability to inhibit the activity of B or T cells. They are used to treat autoimmune diseases caused by an overreaction of immune cells, and unlike conventional autoimmune disease treatments, they do not target just B or T cells. This dual mechanism of action that targets both cells simultaneously is expected to make autoimmune diseases more effective. In addition to Gilead, other BTLA agonists currently in development include ANB-032 from Anaptysbio in the U.S. and HFB-200603 from HiFiBiO Therapeutics in France. Eli Lilly discontinued its Phase 2 clinical trial for lupus, an autoimmune disease, due to the lack of efficacy of its BTLA agonist antibody. Lilly had hoped that activating BTLA to target autoimmune diseases would be a novel approach, but the company abandoned the program after failing to achieve clinical success in psoriasis and lupus.
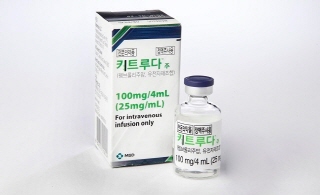
- Policy
- Keytruda’s reimb for gastric cancer will be redeliberated
- by Lee, Tak-Sun Dec 20, 2024 05:46am
- The application to expand the gastric cancer indication for the immuno-oncology drug Keytruda (pembrolizumab, MSD) has been given a ‘redliberation’ decision. The drug is currently seeking reimbursement for 17 indications, but financial issues have prevented a conclusion. The Health Insurance Review and Assessment Service’s Cancer Disease Deliberation Committee held its 9th meeting for 2024 on the 18th and deliberated on the proposed reimbursement standards for anticancer drugs. Keytruda’s result was the center of attention. MSD, the maker of Keytruda, has applied for reimbursement expansion for 17 indications. The CDDC had deliberated Keytruda’s agenda 4 times but deferred the decision due to the submission of an additional financial sharing plan. It was hoped that MSD would present its financial plan and reach a new conclusion, but the outcome was the same. The 9th CDDC meeting deliberated on 2 recent applications for gastric cancer indications for Keytruda but decided to redeliberate the case without reaching a conclusion. On the other hand, expanded reimbursement standards were successfully set for Zytiga (abiraterone acetate, Janssen) and Lorviqua (lorlatinib, Pfizer). The coinsurance rate for Zytiga was reduced from 30% to 5% as a treatment of asymptomatic or mildly symptomatic metastatic castration-resistant prostate cancer reduction from 30% to 5%, and reimbursement standards were set for Lorviqua as a treatment for adult patients with anaplastic lymphoma kinase (ALK)-positive metastatic non-small cell lung cancer (NSCLC). On the new drug front, the lymphoma drug Jaypirca (pirtobrutinib, Lilly) achieved reimbursement coverage. Roche's Columvi (glofitamab) and AbbVie's Epkinly (epcoritamab) failed to set reimbursement standards.

- Policy
- Basic refund type-drugs granted early RSA termination
- by Lee, Tak-Sun Dec 20, 2024 05:46am
- Only drugs categorized as a basic refund type are possible when a pharmaceutical company wishes to terminate the Risk Sharing Agreement (RSA) early. However, pharmaceuticals on the cost-effectiveness evaluation waiver track, demonstrating cost-effectiveness, can be submitted for negotiation through the renewal of the agreement. The National Health Insurance Service (NHIS) has finalized the 'Guidelines for Drug Price Negotiations and Detailed matters.' On the afternoon of December 19, the NHIS held a briefing session at the Korea Pharmaceutical and Bio-Pharma Manufacturers Association (KBPMA) to explain the revisions made to the 'Guidelines for Drug Price Negotiations' and the 'Guidelines for Drug Price Negotiations and Detailed matters.' The finalized revised guidelines include the changes reported to the Health Insurance Policy Review Committee meeting held in December 2024. These changes are part of the 'Proposal to reflect the new drug's innovative value and the nation's healthcare security.' Health Insurance Review and Assessment Service (HIRA) included these updates in the 'Specific evaluation criteria of new drugs and medicines in consideration for negotiation' during revisions made in August and December. Meanwhile, the Ministry of Health and Welfare (MOHW) incorporated these changes in the October administrative notice outlining partial revision to the drug reimbursement and adjustments criteria. The NHIS has improved its policies to align with regulatory revisions. The revised guidelines for drug price negotiations and detailed matters for RSA underwent consultation starting on November 28 and concluded on December 4. On the afternoon of December 19, the NHIS held a briefing session at the Korea Pharmaceutical and Bio-Pharma Manufacturers Association (KBPMA) to explain the revisions made to the Despite differing opinions from some associations, the revisions were finalized as initially proposed. One association suggested that early termination should be allowed for all RSA types. However, the final decision limited early termination to basic refund RSA type only. Current policy states that companies can request early termination of an RSA, but under the revised guidelines, this will be limited to basic refund RSAs. However, for cost-effectiveness-evaluated drugs that have demonstrated cost-effectiveness through the HIRA’s Drug Reimbursement Evaluation Committee (DREC) review, companies, and NHIS can renegotiate to adjust reimbursement ceilings, reset expected claim amounts, or modify·terminate total expenditure-capped RSAs. This opens the possibility of terminating total expenditure-capped RSAs through renewal of agreement. Additionally, one association suggested simplifying re-renewal processes for all RSA types but was rejected. The revision will apply only to basic refund RSAs, as this approach aligns with existing HIRA regulations, which NHIS incorporated into its guidelines. The negotiation period for RSA renewals following contract expiration will also be reduced. If renewal or termination negotiations fail, the NHIS and the company can renegotiate once under the Minister of Health and Welfare’s directive. The drug is removed from the reimbursement list if this second negotiation fails. However, based on the revised guidelines, in case of a negotiation failure, the NHIS and the company will have 60 days for renegotiation under the directive of the Minister of Health and Welfare without any extensions. If renegotiation fails, the drug will be removed from the reimbursement list. "Currently, negotiations can take up to 300 days, which burdens the individuals involved," Oh Se-rim, head of the HIRA's Pharmaceutical Benefits Department, stated. "The revised guidelines reduce this period to a maximum of 240 days." The updated proposal indicates that when applying for reimbursement listings of the same formulation under a RSA, the NHIS and associated organizations may mandate the applicant to sign a confidentiality agreement and provide details regarding the RSA associated with the drug. The recent revision of pricing negotiation guidelines remains intact. Updates include a 30-day timeframe for essential medicines. Additionally, a new provision permits 'pre-negotiations' to occur during the evaluation period conducted by the HIRA. The NHIS has already implemented this strategy in negotiations concerning drugs experiencing shortages, such as acetaminophen, after the COVID-19 endemic.
- Policy
- Cinacalcet impurities spread…Huons recalls product
- by Lee, Hye-Kyung Dec 19, 2024 05:53am
- The detection of excess 'N-nitroso cinacalcet' impurity has led to the recall of the kidney drug cinacalcet hydrochloride. The Ministry of Food and Drug Safety announced on the 17th that Huons’ Calcepara Tab 25 mg (cinacalcet hydrochloride) with 3 manufacturing numbers TTB201 (2025-08-16), TTB301 (2026-05-18), and TTB302 (2026-05-21) will be recalled by the manufacturer. The cinacalcet hydrochloride-based formulations are used for the treatment of secondary hyperparathyroidism in patients with chronic kidney failure undergoing dialysis, and the original drug product, Regpara Tab (cinacalcet hydrochloride), was first recalled on November 20th. List of approved cinacalcet drugs In early October, an Indian drug manufacturer decided to recall the drug due to the detection of nitrosamine impurities in the cinacalcet ingredient, raising the possibility of a recall in Korea. In early November, the U.S. Food and Drug Administration (FDA) announced a voluntary recall on its website after nitrosamines were found to exceed the FDA's recommended daily allowable intake limit. In Korea, Kyowa Kirin Korea recalled some lot numbers of two products, Regpara Tab 25mg and 75mg, due to excessive impurities detected in stability tests. The source of the N-nitroso cinacalcet is believed to be the reaction between the raw materials and excipients. Regpara Tab was launched in Korea in 2011 and became popular as soon as it was launched because it induces a decrease in parathyroid hormone (PTH) while reducing serum phosphorus (P), serum calcium (Ca), and Calcium-phosphorus product (CaxP) levels and it is easy to take once a day without the side effects caused by vitamin D preparations. In particular, generics have been entering the market at a rapid pace since the patent expired in 2017, and there are currently 11 generics approved for cinacalcet in Korea. According to the import and manufacturing performance of drugs disclosed by the Ministry of Food and Drug Safety, 2 doses of the original, Regpara, posted sales of KRW 5.8 billion at the current exchange rate. Alvogen Korea's ‘Cinacet Tab,’ Daewon Pharmaceutical's Repatzin Tab,’ and Yuyu Pharmaceutical's ‘Beneph Tab’ have posted figures of more than KRW 1 billion, while Calcepara Tab, which has been recalled, has posted nearly KWR 143.1 million. An MFDS official said, “Since impurities were detected in ingredients, excipients, and other components, we asked generic drug manufacturers to test whether excess N-nitroso cinacalcet were detected. We plan to consider necessary measures based on the results.” In response to the manufacturer's request for temporary ease of the tolerance standard and the opinions of medical expert organizations on its medical necessity, the MFDS will reportedly take measures such as temporarily raising the impurity threshold. However, manufacturers will be required to notify the agency within a month and implement a countermeasure plan for reducing impurities within 3 years. Meanwhile, Huons is conducting research to ensure that products exceeding the impurity standard are not distributed. It is considering various measures such as shortening the use period and removing or reducing impurities. “We are conducting various studies to reduce the risk of impurities, such as reduction measures,” said a Huons representative, ”and we will complete the research and development faster than the specified period so as not to disrupt the supply for patients.”
- Policy
- Sanofi signs PVA negotiations for hemophilia drug 'Alprolix'
- by Lee, Tak-Sun Dec 19, 2024 05:52am
- Product photo of Alprolix The ceiling price for Sanofi's 'Alprolix,' a new treatment for hemophilia B, is expected to decrease through price-volume agreement (PVA) negotiations. This drug was approved in May 2017, and reimbursement listed in June of the following year. According to industry sources on December 18, the National Health Insurance Service (NHIS) and Sanofi-Aventis Korea agreed on Alprolix's PVA negotiations (TYPE-NA). Alprolix has been considered for the PVA negotiation for the first time after it was reimbursement listed in June 2018. The TYPE-NA negotiation is conducted when the ceiling price is adjusted by the TYPE-GA price or when the bill amount of the same product has increased by more than 60% or 10% from the previous year's bill, and the increase is more than KRW 5 billion. The current negotiation was possible because Alprolix's ceiling price had not been adjusted due to TYPE-GA negotiation, and it met the criteria for TYPE-NA negotiation four years after the first listing. This biological drug received approval in May 2017 as a daily preventive therapy to suppress and prevent bleeding, manage pre- and post-operative care, and reduce the frequency of bleeding in hemophilia B. The pharmaceutical company accepted price 100% below the weighted average price of substitute medicines and was exempted from undergoing the drug negotiation process. It only negotiated for the expected claim amount and successfully listed for reimbursement in June 2018. The ceiling price is KRW 1,181. Meanwhile, the Swedish biotechnology group Sobi owns Alprolix's global sales rights. Sobi partners with Sanofi to sell Alprolix. Sobi established a joint venture with Handok in South Korea in April, named 'Sobi-Handok.'
- Policy
- Hyperphosphatemia treatments undergo generation change
- by Lee, Tak-Sun Dec 19, 2024 05:52am
- Drugs that improve hyperphosphatemia in patients with chronic kidney disease are undergoing a generation change in the domestic market. Following the launch of the new drug Nephoxil Cap (ferric citrate, Kyowa Kirin Korea) last year, generic drugs containing sevelamer have continued to grow, and news of the withdrawal of existing drugs is also arising. According to industry sources on the 18th, Fosrenol Tab (lanthanum carbonate), which was supplied by JW Pharmaceuticals in Korea, will be discontinued. Fosrenol has been imported and sold by JW Pharmaceuticals from Takeda. With the termination of the supply contract, its supply to the domestic market is expected to be gradually discontinued by February next year. Fosrenol is a non-calcium phosphate binding agent that was approved in Korea in January 2006. It formed a three-way tie with Renvela (Sanofi) and Invela (SK Chemical) as a treatment for hyperphosphatemia in 2022. However, Fosrenol’s competitiveness weakened as generic drugs containing sevelamer carbonate, such as Renvela and Invela, were released to the market in July 2022, and the new drug Nephoxil Cap was launched last year. Last year, sales of Renvela, Invela, and Fosrenol all declined. Renvela’s sales fell to KRW 9.2 billion in 2023, down 4% from the previous year, and Invela’s sales fell to KRW 7.4, down 15%. Fosrenol's sales were flat at KRW 4.1 billion ($4.1 billion), the same as the previous year. The emergence of sevelamer generics and new drugs are disrupting the existing market structure. Nephoxil, which was launched last year, is a drug for hyperphosphatemia in chronic kidney disease patients undergoing hemodialysis and is particularly praised for lowering the risk of side effects such as hypercalcemia and vascular calcification of calcium-based binders with iron-based binders. In particular, Kyowa Kirin Korea, which supplies the drug, not only succeeded in getting the drug on the reimbursement list within a year by accepting the weighted average price of KRW 377, which is 90% of the weighted average price of alternative drugs, showing competitivity in drug price. Not only new drugs but also the highest-selling sevelamer drug has seen its number of tablets increase to 9 due to the emergence of generics. This situation explains why Fosrenol, which showed stagnant sales, eventually withdrew from the domestic market. On the 5th, 3 Fosrenol products also withdrew their domestic marketing authorizations. “The market for hyperphosphatemia treatments has recently seen intensified competition as non-calcium-based drugs with fewer side effects have gained prominence due to expanded reimbursement,” said a pharmaceutical industry insider. ”In addition, the introduction of new drugs has weakened the competitiveness of existing products.”
- Policy
- Prior notification of drug permit changes extended to 2025
- by Lee, Hye-Kyung Dec 19, 2024 05:52am
- The operation of the pilot program, 'Advance Notification System for Drug Change Permit', which allows drug manufacturers and importers to apply for change permits on their preferred date, will be extended. According to industry sources on the 18th, the Ministry of Food and Drug Safety (MFDS) will extend the pilot project until December 31 next year to collect sufficient data to evaluate the system and in consideration of industry demand. The Advance Notification System for Drug Change Permit is a system in which the MFDS and drug manufacturer/importer discuss the date of a drug’s permit change in advance, taking into account the manufacturing and import schedule of the company before processing the change, and then approving the change according to the applicant's desired date. The system had been operated only for new drugs, orphan drugs, and advanced biopharmaceuticals until now. Still, as of May 30, the system had been expanded to include drugs subject to production, import, and supply interruption reports. In the case of drugs subject to production, import, and supply interruption reports, their inclusion in the system may be subject to change according to the list announced by the Health Insurance Review and Assessment Service in late December. Drug manufacturers and importers must obtain a change permit from the head of the MFDS when changes are made to authorized drugs, and then manufacture and import products reflecting the changes. Previously, when the MFDS’s approval process for a drug permit change application was completed, its approval was processed without any notification, making it difficult for companies to predict the date of approval. To address this, the MFDS has established an advance notification system for drug permit changes and plans to institutionalize it through evaluation after completion of the pilot program. The system allows a company that applies for a change permit and enter a notice of the desired change period into the NEdrug system after the MFDS completes review. The change date must be set after the statutory deadline, and extensions can be requested within the statutory period of the civil complaint. Adjustment of the date of change approval through the advance notification system is only applicable to cases where it is necessary to process (extend) the period later than the original processing period due to the manufacturing and import schedule of the drug in question in order not to affect the existing approval process.