- LOGIN
- MemberShip
- 2026-06-21 13:47:31

- Company
- Revisions to the guidelines for atopic dermatitis treatment
- by Hwang, Byung-woo Aug 08, 2024 09:25am
- As the market for atopic dermatitis rapidly shifts due to the introduction of new drugs, domestic guidelines are being revised in nine years, and therapeutic guidelines are also changing. The guidelines now suggest a higher recommendation grade for treatments, including biological agents and JAK inhibitors, for use in patients with moderate-to-severe symptoms. They also include detailed recommendations to solve the issue of replacement therapy, which experts focus on. (Clockwise from upper left) Product photos of Dupixent, Rinvoq, Olumiant, Adtralza, and Cibinqo. The Korea Atopic Dermatitis Association (KADA) announced on July 31st the '2024 Guidelines for the Treatment of Atopic Dermatitis in Korea.' This guidelines have been revised after nine years, since 2015. Biological agents (injectable medicines), such as Dupixent (dupilumab, approved Oct. 2018), and JAK inhibitors, such as Olumiant (baricitinib, approved May 2021), have completely changed the paradigm of the treatment for atopic dermatitis. Changes in therapeutic strategy have been reflected in the guidelines following the introduction of new medicines, which can now be prescribed with less economic burden due to reimbursement coverage. The degree of recommendation for biological agents depends on domestic approval. Dupixent and Adtralza (tralokinumab, approved August 2023) were rated as Grade A, the highest recommendation, for treating patients with moderate or severe symptoms that cannot be treated with topical therapy. The basis for a high recommendation grade for these two medicines was based on having real-world studies aligning with clinically shown effects. Considering this factor, medicines that have yet to receive domestic approval, such as lebrikizumab and nemolizumab, were rated as Grade B. Additionally, JAK inhibitors that received domestic approval for treating atopic dermatitis, including Olumiant, Rinvoq (upadacitinib, approved Oct. 2021), and Cibinqo (abrocitinib, approved Nov. 2021), were all rated as Grade A. However, the recommendation was based on the premise that Olumiant and Rinvoq, which also have indications to treat inflammatory diseases, may be more effective than Cibinqo, which is indicated only to treat atopic dermatitis. Furthermore, unlike biological agents, JAK inhibitors were granted a Grade A recommendation based on conducting early diagnostic monitoring testing. The KADA emphasized that patients be tested for hemocyte count and kidney and liver function four weeks after the treatment for monitoring, and re-assessment every three months during the treatment. The KADA emphasizes replacement therapy…"should be allowed to be used when there are insufficient medicines" Despite the growing number of atopic dermatitis treatment options, clinical practices advocate lifting restrictions on replacement therapy. The issue arises because replacement therapy is authorized for psoriasis within the same dermatology practice. Therefore, itt has been included in the revised guidelines. The main point of the argument is that patients with atopic dermatitis may have different responses. Although biological agents may be effective in treating moderate-to-severe atopic dermatitis, some patients may not respond well to the treatment. Insufficient response, as defined by the KADA, is 'Meeting one or more criteria of having not reached 50% in Eczema Area and Severity Index (EASI) score, suffering from daytime or nighttime pruritus with NRS score ≥4, or having Dermatology Life Quality Index (DLQI) ≥6. The KADA explained, "There are no useful biomarkers to predict the treatment outcomes of biological agents or JAK inhibitors for atopic dermatitis treatment." They added, "Currently, when patients with moderate atopic dermatitis do not respond well or have adverse reactions to the treatment with biological agents or JAK inhibitors, they cannot switch to other medicines for effective treatment." However, the revised guidelines have not specified which biological agents and JAK inhibitors can be used for replacement therapy. Some studies suggest concrete clinical evidence for replacement therapy, but studies with low-grade evidence exist. The KADA stresses that it will not set a hurdle for replacement therapy between biological agents and JAK inhibitors when patients do not respond well to initial treatments. During a press conference, Professor Ahn Ji Young at the National Medical Center, stated, "The sequence for replacement therapy has not been determined as we have not yet concluded the appropriate medicines for this purpose. The KADA hopes that replacement therapy will be possible regardless of types of medicines used." According to the pharmaceutical industry, unlike the initial request for authorization of replacement therapy, the government may be more willing to bring changes now. For instance, the Health Insurance Review and Assessment Service (HIRA) has recently requested supplementary documents. Therefore, replacement therapy may be considered based on KADA's suggestion for patients with insufficient response. An industry official said, "The revised guidelines in South Korea and other countries, such as the United States and Europe, now include various recommendations and evidence for replacement therapy when a patient fails initial treatment." He added, "We cannot predict an accurate date for change, but we hope that accumulating evidence will be used toward opening positive discussion."

- Company
- RSV vaccine market for children is expected to be big
- by Hwang, Byung-woo Aug 08, 2024 09:24am
- As Sanofi is set to launch its injectable antibody drug to prevent respiratory syncytial virus (RSV) lower respiratory tract disease for the first time in South Korea, the company begins marketing. The company aims to raise awareness of RSV disease to promote vaccination. Sanofi has strategized a top-down approach, starting with labor and delivery hospitals, and expanding to private practices. As clinical practices have high demand for the vaccine, it is likely to enter the market quickly. Product photo of Beyfortus According to industry sources on August 6th, Sanofi is set to launch Beyfortus, an injectable antibody for RSV prevention, ahead of the upcoming 2024-2025 vaccination season. Beyfortus received approval from the Ministry of Food and Drug Safety (MFDS) in May. It is an injectable antibody to prevent lower respiratory tract diseases, including pediatric pneumonia and bronchiolitis. Previously, RSV immunization product for infants and children in South Korea was only available for high-risk infants and children who are expected to have a high risk of contracting severe RSV disease, including preterm infants. However, Beyfortus can be treated in all infants and children. The RSV vaccination season in South Korea typically lasts from October to March, when RSV spreads. In other words, Sanofi must devise a market entry strategy since it must launch soon. Fortunately, the market is receptive. RSV in children is typically regarded equivalent to influenza in children contracted globally during a particular season. An executive from the Korean Society of Pediatric Infectious Diseases explained, "RSV infection rate is extremely high with 50-60% of children are infected by RSV within the first 1-3 years of life, and 100% of children get RSV by their third birthday." He added, "Once infected, the symptoms are unlike the common cold. Therefore, the expenses and caregivers support are burdensome." After the launch, Beyfortus vaccination will be non-reimbursed. However, the early vaccination rate is expected to be high, considering that the market for infants and children is based on demand for vaccination rather than cost. 사노피는 지난 달 말부터 RSV 캠페인을 진행중이다 Ahead of the upcoming launch, Sanofi puts efforts into raising awareness of RSV disease. For example, Sanofi has started an 'It turned out to be RSV, story contest,' aiming to gather patient stories related to the disease and share them. Park Hee-kyung, President of Vaccine Division at Sanofi-Aventis Korea, explained, "Because RSV virus is a major cause of infant and children hospitalization, it is crucial for parents to catch early symptoms and respond to them. I hope the contest will provide an opportunity to raise awareness of RSV infection-related symptoms and lower respiratory tract disease." The vaccine has been already introduced to national immunization programs in other countries, including the United States, and real-world effects have been reported. Consequently, Sanofi aims to provide vaccine-related information to parents by working together with clinical institutes. Then, how will the Beyfortus immunization provided? Although the type of drug differs, the vaccination is expected to begin in big hospitals and expand to private clinical practices, similar to the case of GSK's shingles vaccine Shingrix. For Shingrix, the vaccination was first provided in university hospitals and general hospitals to immune-compromised individuals who previously could not get shingles vaccination. Sanofi official said, "Beyfortus is recommended to be administered to infants at birth for those born during the RSV season, or before they are discharged." One added, "Children born before the beginning of the RSV season are recommend to be administered before the start of the season during their visit to the pediatrics hospital for regular immunization." Typically, when a vaccine is not part of the essential immunization program, individuals are likely to consider vaccination during their hospital visits. Beyfortus, which is not a vaccine but provides similar benefits, immunization is likely to begin in labor and delivery hospitals, where individuals are provided information on diseases and products. Division of Infectious Diseases professor at a university hospital in Seoul said, "Because newborns, infants, and children are most vulnerable to RSV, it is a good option for providing preventative effects." Professor added, "Immunization is expected to increase gradually rather than robustly, considering the disease awareness."

- Company
- Celltrion presents clinical results of Prolia biosimilar
- by Hwang, Byung-woo Aug 08, 2024 09:24am
- Celltrion announced on August 6th that it has published the global Phase 3 clinical trial results evaluating the efficacy and safety of CT-P41, a biosimilar referencing the osteoporosis drug Prolia (ingredient: denosumab). The Phase 3 study compared the safety profile, including the efficacy, pharmacodynamics, pharmacokinetics, and immunogenicity, of CT-P41 to the original drug, in 479 female patients with osteoporosis after menopause. The study was conducted in four European countries. The results showed that at a 78-week evaluation, the primary endpoints of the efficacy and pharmacodynamics between CT-P41 and the original drug met the equivalence criteria. Additionally, the efficacy and safety of CT-P41 have been demonstrated in patients who received the drug 52 weeks after the initial treatment with the original drug. As the primary endpoint, lumbar spine bone density changes from baseline to 52 weeks in both the CT-P41 and the original drug treatment groups were assessed. The results showed that the differences between the two groups met predefined equivalence criteria. The study also demonstrated equivalence through the area under the curve (AUC) for the primary pharmacodynamic endpoint, the bone metabolism marker 'bone resorption marker (s-CTX),' over the first six months. Prolia is an osteoporosis treatment, and the same active ingredient received approval under the product name of 'Xgeva,' a drug used to prevent bone metastasis complications in cancer patients. The drug's global sales amounted to approximately US$6.16 billion (about KRW 8 trillion). Celltrion official said, "As we have confirmed the efficacy and safety of CT-P41 compared to the original drug, we will strive to proceed with ongoing approval applications in major global countries." He added, "We plan to expand our treatment portfolio quickly in various fields, including bone disease, eye disease, and allergy disease, to enhance the company's growth." The study results have been published in the 'Osteoporosis International,' the official journal of the International Osteoporosis Foundation (IOF) and the Bone Health & Osteoporosis Foundation (BHOF).
- Company
- Qalsody receives orphan drug designation in Korea
- by Eo, Yun-Ho Aug 08, 2024 09:24am
- ‘Qalsody (tofersen),’ a new drug for Lou Gehrig's disease, was designated as an orphan drug in Korea. The Ministry of Food and Drug Safety (MFDS) recently announced the designation through an orphan drug designation notice. More specifically, the drug is indicated for amyotrophic lateral sclerosis (Lou Gehrig's disease) associated with a mutation in the SOD1 (Superoxide Dismutase 1) gene. Qalsody, which was developed by Biogen, is an antisense oligonucleotide (ASO) drug that blocks the messenger ribonucleic acid (mRNA) associated with the SOD1 gene mutation to prevent its expression. The drug was approved by the U.S. FDA in June last year, followed by the European EMA in May. The efficacy data for Qalsody is not exactly glowing. However, this is likely a reflection of the scarcity of treatment options for the disease. In the Phase III VALOR trial, Qalsody failed to meet its primary endpoint, ‘Amyotrophic Lateral Sclerosis Functional Rating Scale (ALSFRS).’ However, it did reduce the secondary endpoints of ‘increased SOD1 protein levels in the cerebrospinal fluid’ and ‘neurofilament light chain (Nfl) concentrations’ by 26-38% and 48-67%, respectively. The most common adverse events reported in the trial were pain (back, arms, legs), fatigue, muscle and joint pain, fever, and increased protein and white blood cell counts in the cerebrospinal fluid. ALS is a rare neurological disease that affects nerve cells in the brain and spinal cord responsible for muscle movement and can lead to progressive paralysis and death. Although a considerable number of clinical trials are being conducted relative to the incidence rate, most medications only provide symptom relief.

- Company
- Vaxneuvance increases presence in NIP mkt
- by Hwang, Byung-woo Aug 08, 2024 09:24am
- MSD Korea is accelerating its efforts to capture the national immunization program (NIP) market by touting the high immunogenicity of Vaxneuvance. The vaccine has already been rapidly introduced to general hospitals and clinics upon its launch, and the company is highlighting the vaccine’s clinical benefits to gain a competitive advantage. Dr. Hyun-mi Kang, Professor of Pediatrics, St. Mary Vaxneuvance, which was approved late last year, is the first new pneumococcal vaccine introduced to Korea in 13 years. It is a 15-valent vaccine that added 2 serotypes -22F and 33F – to the existing 13-valent vaccine. In particular, the vaccine made the pediatric NIP list at an unprecedented speed, just one month after its approval. Since its launch in April, Vaxneuvance has been available for NIP vaccination in children aged 2 months to 5 years. Previously, Pfizer's Prevenar 13 was the only available NIP vaccine for pediatric pneumococcal disease. Now, MSD is expanding Vaxneuvance’s presence in the market by emphasizing that children who have received one or more doses of Prevenar 13 can also cross-vaccinate the remaining doses with Vaxneuvance. At the Vaxneuvance media seminar that was held on June 6, experts said that the introduction of Vaxneuvance has changed clinical practice. "We don't know the overall vaccination rate because we don't have specific data, in my hospital, Vaxneuvance is being administered first due to its broader coverage since April," said Hyun-mi Kang, professor of Pediatrics at St. Mary's Hospital in Seoul. Jaeyong Cho, Executive Director of MSD Korea's Vaccine Business Unit, added, "Although the data is unofficial, there has been an increase in Vaxneuvance’s use as third and fourth doses along with the initial dose since its launch. Overall, the rate of those receiving an initial dose of Vaxneuvance is higher, but the proportion of cross-vaccinations is also continuing to grow." Vaxneuvance’s strength is in the high immunogenicity of all serotypes Immunogenicity was also highlighted as the competitive advantage of Vaxneuvance during the media seminar. This means that there is a need to choose a highly immunogenic option to prevent invasive pneumococcal disease (IPD), which is highly fatal in children. The WHO's definition of immunogenicity is "the ability of a vaccine to induce a measurable immune response. The specific serotype-specific immunogenicity criteria are 'IgG concentration of 0.35 μg/mL or higher'. "In a Phase III study in healthy Korean infants and young children, Vaxneuvance demonstrated a primary immunogenicity endpoint of IgG of 0.35 μg/mL or higher across 15 serotypes after the 3rd dose in more than 95% of subjects," said Professor Kang. While Vaxneuvance is rapidly penetrating the market based on its clinical performance, there is hesitation in the field as Prevenar 20, which contains more serotypes, will be launched in the second half of the year. In this regard, Professor Kang emphasized that it is also important to consider the prevention effect of each serogroup in addition to the broader spectrum of protection. "We have been saying that preventing more is important for invasive pneumococcal disease due to its many serotypes, but the more number of serotypes does not necessarily mean that (the vaccine) is good. I think we need to consider the safety of the vaccine along with the immunogenicity for each serotype."
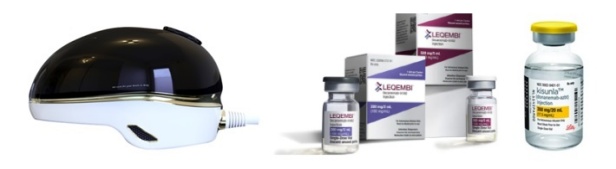
- Company
- Electronic drugs show potential to conquering Alzheimer's
- by Son, Hyung-Min Aug 08, 2024 09:24am
- The Korean pharmaceutical bio industry is making a bid into the electronic drug market for Alzheimer's disease. Recently, Remed unveiled the results of its transcranial magnetic stimulation (TMS) therapy that demonstrated an effect in treating Alzheimer's disease. AriBio, which is developing a new drug for Alzheimer's disease, is developing an electronic drug that uses vibroacoustic stimulation On July 7, Remed disclosed clinical results showing that an electronic drug being developed by Remed was effective in patients with Alzheimer's disease. Remed is a Korean biotech company that has been identifying the potential of electronic drugs in various areas including depression, attention deficit hyperactivity disorder (ADHD), and dementia. The electronic drug being developed by Remed for Alzheimer's disease uses transcranial magnetic stimulation. Transcranial magnetic stimulation is a brain stimulation procedure that uses magnetic fields to stabilize or activate brain nerve cells in a specific area at the base of the skull. It has been used to treat various brain disorders such as depression, intractable obsessive-compulsive disorder (OCD), and chronic pain. Remed’s subsidiary NextCure’s helmet-type wearable electronic therapyRemed has confirmed a change in the Alzheimer's Disease Assessment Scale-Cognitive Subscale (ADAS-Cog), an important indicator in the diagnosis of dementia, with its electronic drug in clinical development. The clinical trial was conducted from May 2020 to April 2022 to evaluate the efficacy and safety of the electronic drug in 30 patients with Alzheimer's disease. Trial results showed that the ADAS-Cog value of patients treated with the electronic drug improved significantly compared to those who did not receive treatment. More specifically, the change in ADAS-Cog values was above the ADAS-Cog MCID threshold of 4.0, which is regarded as a clinically significant change in dementia medications, and showed no serious adverse events. "These results suggest that transcranial magnetic stimulation may be effective in Alzheimer's patients in addition to depression, intractable OCD, chronic pain, and stroke," said the researchers involved in the trial. Remed plans to complete the exploratory trial and submit a confirmatory IND to the Ministry of Food and Drug Safety. AT&C is developing a treatment system that combines an electronic drug that uses transcranial magnetic stimulation and digital therapeutics (DTx) for the treatment of mild Alzheimer's disease. The company is currently conducting a confirmatory clinical trial with 158 patients at Korea University Anam Hospital, Dong-A University Hospital, Chonnam National University Hospital, Jeonbuk National University Hospital, Chung-Ang University Gwangmyeong Hospital, Chungnam National University Hospital, and Hanyang University Hospital. According to the company, the combination of transcranial magnetic stimulation and digital therapeutics showed better and longer treatment effects than conventional medication and cognitive training, which showed low treatment effects and short treatment duration. In addition to Remed and AT&C, another company, AriBio, is developing electronic drugs for Alzheimer's disease. In May, Aribio received approval from the Ministry of Food and Drug Safety to conduct a clinical trial for an electronic drug that uses vibroacoustic stimulation on Alzheimer's disease patients. The company’s brain vibroacoustic stimulation electronic drug offers a new approach to treating Alzheimer's disease. The company explained how their trial is the first clinical trial of an electroacoustic brain stimulation device that uses vibroacoustic stimulation, although there have been devices that use electric, electromagnetic field, and ultrasonic methods in the past. The clinical trial will be conducted by Professor Sang-Yoon Kim’s team from the Seoul National University Bundang Hospital, to confirm the efficacy and safety of brain vibroacoustic stimulation. The efficacy of the electronic drug will be evaluated in a double-blind, 6-month trial in 30 patients with early-stage Alzheimer's disease, including those with mild cognitive impairment. If the exploratory clinical trial confirms the efficacy of the electronic drug, AriBio aims to launch the electronic drug for dementia by 2026 after conducting a confirmatory clinical trial. In addition to the electronic drug, AbiBio is also conducting a Phase III clinical trial on AR1001, a novel drug candidate for Alzheimer's disease. AR1001 targets multiple causes of Alzheimer's disease, including the PDE5 protein. Recently, studies have shown that phosphodiesterase 5 (PDE5) inhibitors, such as Viagra and Cialis, are effective in preventing Alzheimer's disease, raising interest in AR1001's commercialization potential. The introduction of electronic drugs in the field...will Alzheimer's disease finally be conquered? Eisai and Biogen’s Alzheimer’s treatmentAlzheimer's disease has been a challenging area for drug development. After the hypothesis that Alzheimer's disease is caused by amyloid beta emerged, targeted drugs were developed, and then were found ineffective. One such drug was Eisai’s Aduhelm (aducanumab). The amyloid beta hypothesis was highly suspect, so there was not much confidence in the drug, but the drug worked. However, Aduhelm was withdrawn from the market due to its high price and concerns about side effects. Then came Leqembi (lecanemab), developed by Eisai and Biogen. Leqembi showed efficacy in early-stage Alzheimer's disease in clinical trials and has cleared regulatory hurdles in South Korea, the U.S., Japan, and China. Lilly’s Alzheimer’s treatmentMore recently, Lilly's Kisunla (donanemab) has also been approved by regulatory authorities. Kisunla was shown to delay cognitive deterioration in patients with early Alzheimer's disease in the Phase III TRAILBLAZER-ALZ2 study. In the study, donanemab delayed cognitive deterioration regardless of disease progression or pathologic stage. However, there is currently no cure for Alzheimer's disease. Although Leqembi and Kisunla have emerged, experts believe that they are only effective in patients with early-stage Alzheimer's disease. This is why the pharmaceutical industry continues to tackle Alzheimer's disease with a variety of possibilities, including not only new drugs but also electronic drugs and digital therapeutics. To date, clinical trials using electronic drugs have been limited to exploratory trials, but if they show effectiveness in confirmatory trials, they may also show synergy with the new drugs on the market.
- Company
- Tepezza receives orphan drug designation in Korea
- by Eo, Yun-Ho Aug 06, 2024 09:16am
- Tepezza (teprotumumab), a targeted treatment for thyroid eye disease (TED), has been designated as an orphan drug in Korea. The Ministry of Food and Drug Safety (MFDS) recently announced the designation through an orphan drug designation notification. Specifically, Tepezza is indicated for the treatment of adult patients with moderate-to-severe thyroid ophthalmopathy. Tepezza is an insulin-like growth factor-1 receptor inhibitor class monoclonal antibody that is administered intravenously every 3 weeks (8 injections in total). The drug was first approved by the U.S. FDA after receiving a fast-track designation in January 2020 but has not yet been commercialized in Korea. The company recently submitted applications for the drug’s approval in Europe and Japan. The drug was originally developed by Irish pharmaceutical company Horizon Therapeutics, and Amgen acquired the rights to the drug when it acquired Horizon in 2022. In the Tepezza trials, patients with TED were administered 8 infusions of Tepezza or placebo every 3 weeks. The Phase II and OPTIC studies included a total of 121 patients with TED with a Clinical Activity Score (CAS) of 4 or greater and TED onset of 9 months or less. Among the 99 patients aged 40 years or older, the mean age was 54.5 years. 72.7% were women and double vision was confirmed in 74.7% of the patients. Among the 22 patients under the age of 40, the mean age of 32.8 years, 59.1% were female, and 68.2% had double vision. When analyzing the response to Tepezza in patients with acute TED, the proportion of patients who responded to treatment with at least a 2mm improvement in ocular protrusion at week 24 was 86.4% in the under-40 group and 79.8% in the over-40 group, showing no statistically significant difference between the two groups. Tepezza has been recognized for changing the treatment landscape for TED. Prior to Tepezza, the only treatment options available for TED were steroids and orbital decompression. Although steroids reduce inflammation, they are associated with side effects and have a risk of recurrence upon discontinuation, and orbital decompression is associated with surgical complications.

- Company
- Lily Korea appoints John Bickel as new General Manager
- by Hwang, Byung-woo Aug 06, 2024 09:16am
- John Bickel, new General Manager of Eli Lilly KoreaEli Lilly Korea announced on August 5th the appointment of John Bickel as the new General Manager of Eli Lilly Korea, effective August 1st. Bickel is an expert with 26 years of extensive leadership experience at Eli Lilly and Company. In the headquarters, he was responsible for the U.S. and global market in oncology and neuroscience divisions, overseeing various pharmaceuticals. Bickel served as the Chief Marketing Officer (CMO) in the Japanese subsidiary of Eli Lilly and Company from 2019. He also served as the Chief Operating Manager (COO) from January 2023, managing Japan's entire marketing and sales organization. Bickel is known to have contributed to strengthening Japanese subsidiary's business capacity, introducing a digital and omnichannel marketing approach, and utilizing data analysis tools to make key decisions. Furthermore, Bickel played a vital role in talent recruitment and training to carry out innovative marketing strategies. "I am pleased to be appointed as the new leader of Eli Lilly Korea and acknowledge the responsibility. In Korea, innovative pharmaceuticals are gaining attention in the fields of diabetes, obesity, cancer, and immune disease, which Eli Lilly has focused on," Bickel said. "As the new GM of Eli Lilly Korea, I will put efforts into quickly supplying Lilly's new pharmaceuticals to South Korea so that Korean patients no longer suffer from life-threatening diseases that lower quality of life and have better daily lives. Furthermore, I will strive to be a trusted partner in contributing to domestic healthcare and the development of pharmaceutical industry," Bickel said. Meanwhile, Bickel graduated with a Bachelor of Pharmacy from Butler University in 1988 and joined Eli Lilly the same year.
- Company
- Trials start for 'cancer vaccine+immunooncology drug’ combo
- by Son, Hyung-Min Aug 06, 2024 09:16am
- Global pharmaceutical companies are making progress in developing messenger ribonucleic acid (mRNA) cancer vaccines and immuno-oncology combinations. Recently, Regeneron and BioNTech confirmed efficacy in a Phase II clinical trial for melanoma. Moderna, a company specializing in the development of mRNA vaccines, has also shown promise in melanoma and head and neck cancer using its vaccine in combination with the immuno-oncology drug Keytruda. In Korea, LG Chem, ImmuneOncia, and others are developing new mRNA and immuno-oncology drug combinations. A cancer vaccine activates the immune system by administering cancer cell antigens to patients, rather than being a preventive concept. Like a vaccine, it has a mechanism that activates the immune response and causes cancer cells to kill themselves. Major pharmaceutical and biotech companies are combining immuno-oncology drugs and antibody-drug conjugates (ADCs) focusing on maximizing the treatment effect rather than cancer prevention to increase their commercialization potential. #i According to industry sources on the 15th, a cancer vaccine and immuno-oncology drug combination using the cancer vaccine developed by the German BioNTech had demonstrated efficacy in a Phase II study in melanoma patients. The trial evaluated the clinical activity of BioNTech's mRNA vaccine BNT111 in combination with Regeneron's PD-1 immuno-oncology drug Libtayo (cemiplimab). BNT111 is a cancer vaccine that enhances T-cell responses to 4 antigens (NY-ESO-1, MAGE-A3, tyrosinase, and TPTE) expressed in melanoma patients. In clinical trials, the BNT111 and Libtayo combination demonstrated a statistically significant improvement in overall response rate (ORR). The specifics will be presented at an upcoming international conference. BioNTech is also currently conducting a Phase II clinical trial comparing the efficacy and safety of another cancer vaccine candidate, Autogene cevumeran, with Roche's immuno-oncology drug Tecentriq (atezolizumab). Autogene cevumeran is a therapeutic personalized cancer vaccine candidate that screens and encodes up to 20 patient-specific cancer mutations that can act as neoantigens. The trial evaluated the efficacy of the Autogene cevumeran + Tecentriq combination with the FOLFIRINOX regimen ((luorouracil+leucovorin+irinotecan+oxaliplatin). In addition to melanoma, Moderna is also exploring the potential of combining its cancer vaccine candidate mRNA-4157 with MSD's immuno-oncology drug Keytruda for patients with head and neck cancer. The company recently announced clinical results from a study on patients with head and neck cancer The trial evaluated the efficacy and safety of Moderna’s candidate mRNA-4157 in combination with MSD's immuno-oncology drug Keytruda (pembrolizumab) in 22 patients with human papillomavirus (HPV)-negative head and neck squamous cell carcinoma. In the trial, the objective response rate (ORR) for the mRNA-4157 plus Keytruda combination was 27%, compared with the 18% achieved in the Keytruda monotherapy arm. Overall survival (OS) for the combination was 24.6 months, which was longer than the 9.8 months achieved in the Keytruda monotherapy arm. Moderna is currently conducting a Phase III clinical trial for the mRNA-4157 plus Keytruda combination in melanoma. In an ongoing Phase II trial in melanoma patients, mRNA-4157 plus Keytruda was shown to reduce the risk of recurrence or death by 49% compared to Keytruda monotherapy. Domestic companies such as Yuhan Corp and ImmuneOncia start developing mRNA+ immuno-oncology drugs# The domestic pharmaceutical bio-industry is also confirming the potential of the mRNA and immuno-oncology combination therapy. Yuhan Corp is developing new mRNA and LNP (lipid nanoparticles) source technologies through industry-academia collaboration. Yuhan Corp is developing mRNA and LNP (lipid nanoparticle) source technologies with the research teams of Professor Hyukjin Lee from Ewha Womans University and Professor Ju Youp Lee from the University of Cincinnati. The company plans to develop a new type of mRNA structure technology that can increase the amount of protein expression while overcoming the easily degradable stability limitations of mRNA. ImmuneOncia is conducting joint research on mRNA-based next-generation immuno-oncology drugs with mRNA therapeutics developer NuclixBio. The two companies will develop next-generation therapeutics using ImmuneOncia’s immuno-oncology drug IMC-001 and NucleixBio's mRNA-based antibody platform 'ringRNA'. The principle is that when mRNA expressing the same amino acid sequence as ImmuneOncia’s antibody is produced and administered, antibodies are produced in the body, providing an effect. Earlier this year, GI Innovation signed a business agreement with ProGen and SL Vaxigen to evaluate the potential of the immuno-oncology drug 'GI-101A' in combination with a cancer vaccine. The three companies will initiate a Phase II clinical trial of GI-101A and DNA vaccine therapy PG-101 in prostate cancer patients. The trial will be led by Prof. Jae-Lyun Lee of the Department of Medical Oncology at Asan Medical Center in Seoul and has received investigator-initiated clinical approval from the Korean Cancer Study Group (KCSG). GI-101A has a mechanism of action that acts on CD80 and interleukin (IL)-2. IL-2 is involved in immune cell proliferation and activation, and CD80 blocks CTLA4, a receptor that inhibits immune cells from attacking cancer cells.
- Company
- Entresto sales soar… rises 22% in 1 year
- by Kim, Jin-Gu Aug 06, 2024 09:15am
- Pic of Entresto Sales of Novartis' heart failure drug 'Entresto' continues to grow over 20% YoY in its 7th year on the market. At this rate, the drug is expected to generate prescription sales of around KRW 70 billion by the end of this year. The variable is the patent dispute. The company is currently awaiting its second judgment on its patent dispute with generic companies. The patent court’s ruling is expected to serve as a turning point for the early release of generics. Entresto in its 7th year of release, continues to grow 22% YoY According to the drug market research firm UBIST on the 5th, Entresto posted prescription sales of KRW 17.1 billion in Q2. This is a 22% YoY increase from the KRW 14 billion it had posted in Q2 last year. Entresto is a first-in-class angiotensin receptor-neprilysin inhibitor (ARNI) that combines the angiotensin receptor blocker (ARB) valsartan and neprilysin inhibitor sacubitril. It was granted reimbursement and released in 2017. Quarterly prescriptions of Entresto (Unit KRW 100million, Source: UBIST)The drug is still showing strong growth in its 7th year of release. Since its launch, its drug price has been reduced by 21%, through 7 price cuts until earlier this year through the price-volume agreement and voluntary reductions, but it did not have a significant impact on the upward sales trend. This is likely due to the fact that the drug's reimbursement coverage has been expanded at the same time as the price cuts. When Entresto was first added to the reimbursement list in 2017, it was reimbursed as a treatment for patients with chronic heart failure with reduced ejection fraction (HFrEF). Coverage was limited to patients previously receiving an ACE inhibitor or ARB inhibitor in combination with standard therapy at a stable dose for at least four weeks. In March 2022, the reimbursement standard was extended to first-line therapy and became available for patients who were previously untreated with an ACE inhibitor or an ARB inhibitor. In July of the following year, the drug became available for prescription in both inpatient and outpatient settings. The drug's longtime dominance of the heart failure treatment market has also contributed to its steady rise in sales. Entresto was the only prescribed treatment targeting heart failure for 6 years until Bayer launched Verquvo (vericiguat) in September last year. Will its sales exceed KRW 70 billion by the end of the year?...the 2nd ruling of the patent dispute remains a variable The industry expects Entresto to generate prescription sales of around KRW 70 billion by the end of the year. Entresto’s sales have grown rapidly since exceeding the KRW 10 billion mark in annual prescription sales in 2019. The drug posted KRW 57.5 billion last year. In the first half of this year, it generated KRW 32.9 billion. If sales continue to grow at this pace, sales could reach KRW 70 billion in annual prescription sales. The variable is in the patent dispute. Domestic pharmaceutical companies have filed patent appeals against several of Entresto's patents at the same time. Since January 2021, generic companies have filed a challenge on Entresto's crystalline form patent, salt and hydrate patent, 2 use patents, and 2 formulation patents. The generic companies won the first trial. After losing the first trial, Novartis appealed 3 cases: the crystalline form patent, the use patent, and the salt and hydrate patent. Of these, the patent court rejected the use patent case in November last year. Novartis appealed again to the Supreme Court, but the court ordered the discontinuance of the trial in April this year. Entresto Patent Dispute Status (Source:KIPO) As a result, 2 patent disputes now remain. The arguments for the salt-hydrate patent have been finalized. The crystalline patent is also reportedly in the final stages of defense. The industry expects the patent court to make a ruling as early as this year. The decision of the second trial is expected to serve as a turning point for early generic launches. Even if Novartis loses the second trial, the company will likely appeal to the Supreme Court, but if generic companies win both the first and the second trial, it will raise the probability of the early launch of Entresto generics.