- LOGIN
- MemberShip
- 2026-06-14 02:56:54

- Company
- Philips Korea's sales staggering…shift to service-centric
- by Hwang, byoung woo Apr 23, 2026 10:51am
- Philips Korea’s sales growth, which had been on a recovery trend since 2023, has stalled, with growth rates remaining in the 1% range.However, during the same period, operating profit increased significantly, showing an actual improvement in profitability. This is interpreted as the result of a shift in the sales mix, moving away from a hardware sales-centric structure toward services, including maintenance and software.Analysis suggests that amid intensifying competition in the large-scale medical equipment market, the future growth is shifting from hardware to software and solutions.Sales Growth 'Stalls,' Operating Profit 'ReboundsAccording to Philips Korea’s recently disclosed audit report, the company’s sales growth appears to have entered a period of stagnation.Specifically, sales, which recorded KRW 336.6 billion in 2021, decreased to KRW 316.1 billion in 2022 before rebounding to KRW 352.9 billion in 2023.Since then, however, sales reached KRW 361.6 billion in 2024 and KRW 364.6 billion in 2025, marking two consecutive years of growth limited to the 1% range following the 2023 rebound, maintaining a slowing growth.One of the primary reasons for this sales stagnation can be traced back to the large-scale business restructuring carried out in 2021. As of September 9, 2021, Philips Korea completed the divestment of its Domestic Appliances division, which was highly profitablePhilips Korea's 5-Year Sales & Operating Profit: BLUE-Sales, RED-Operating profit (source: audit report, unit: KRW 100 million)This was part of Philips’ global 'Selection and Concentration' strategy to focus on the healthcare B2B (business-to-business) sector.Considering that the Domestic Appliances division generated approximately KRW 41.8 billion in annual sales at the time, the separation of this core consumer cash cow is interpreted as a factor in the reduction of the company's overall scale.In contrast to the slowdown in top-line growth, profitability improved significantly. Philips Korea’s operating profit shifted from KRW 42.7 billion in 2021 to a loss of KRW -4.9 billion in 2022.After successfully returning to a surplus of KRW 1.8 billion in 2023, profit showed a recovery to KRW 7 billion in 2024, followed by KRW 36.4 billion in 2025, a more than five-fold improvement compared to the previous year.This indicates that focusing on internal stability and reorganizing the portfolio toward high-value-added businesses, rather than external expansion, has proven effective.Philips Diversifies the Profit Structure, Shift From Product Sales to 'Service'The primary reasons for the improvement in operating profit are cost reductions and lower selling, general, and administrative (SG&A) expenses.The cost of goods sold, which was KRW 270.7 billion in 2024, decreased to KRW 246.3 billion in 2025, and SG&A expenses also fell by approximately 2 billion KRW, from 83.8 billion KRW to KRW 81.8 billion. Essentially, while sales barely grew, profit rose by simultaneously reducing costs and expenses.Furthermore, the fact that the sales structure is increasing 'service sales,' which provides maintenance and software-linked solutions in addition to equipment delivery, appears to have influenced the improvement in profitability.Looking at Philips Korea’s sales structure by business segment (item), product sales decreased from KRW 242.9 billion in 2021 to KRW 219.6 billion in 2022, before recovering to ▲KRW 249.9 billion in 2023 and ▲KRW 254.6 billion in 2024.However, in 2025, product sales recorded KRW 253.6 billion, showing negative growth compared to the previous year.In contrast, service sales has been on a steady upward trajectory, with no negative growth. Service sales, which was KRW 93.7 billion in 2021, recorded KRW 96.5 billion in 2022, ▲KRW 102.9 billion in 2023 ▲KRW 106.9 billion in 2024 ▲KRW 110.9 billion in 2025.The share of service sales in total sales also expanded from 27.8% in 2021 to approximately 30.4% in 2025.Considering the characteristics of the Korean market, where replacement cycles for large medical equipment (MRI, CT, ultrasound, etc.) are long and large-scale orders following the establishment of new hospitals are limited, the expansion of service-oriented sales, such as software upgrades and Service Level Agreements (SLA) for pre-installed equipment, is analyzed to have played a key role in defending profitability.Philips is expanding its cooperation by signing smart hospital business agreements with major hospitals.Intensifying Imaging Competition… The Challenge of Growth DriversBecause of this, Philips Korea is also seeking a breakthrough by creating a digital healthcare environment centered on artificial intelligence (AI) technology.As global competitors such as Siemens Healthineers and GE HealthCare integrate AI solutions with hardware, Philips is moving to capture the early market.For example, Philips Korea is establishing smart hospital collaborations focused on AI-based improvements to ultrasound and imaging workflows.With the recent domestic medical environment emphasizing smart hospitals amid personnel shortages, Philips appears focused on embedding its portfolio, spanning diagnostic imaging·ultrasound·interventional procedure systems, into these systems.However, expanding the influence of diagnostic imaging equipment, which is the most fundamental driver of external growth, remains a challenge.In this context, Philips' next-generation spectral CT, the Verida system, which recently received U.S. Food and Drug Administration (FDA) clearance, could potentially provide a way forward.The core of Verida system is its spectral technology. While most CT scanners focus on providing structural information by imaging the human body at a single energy, spectral CT analyzes differences in tissue composition across multiple energy spectra.As it promotes clinical efficiency improvement rather than just simple image quality enhancement, there is potential for synergy with future smart hospital system construction.An official from the medical device industry stated, "While Philips Korea's new products and technological innovations, such as helium-free MRI and cardiac ultrasound, are receiving positive responses, hospital investment decisions are made on a mid-to-long-term basis due to the nature of the market," and added, "The impact of new products is more likely to be reflected in mid-to-long-term trends rather than short-term performance."
- Company
- Will Perjeta be reimbursed as postoperative adjuvant therapy?
- by Eo, Yun-Ho Apr 23, 2026 10:50am
- Insurance reimbursement criteria for the breast cancer drug Perjeta may be expanded to cover its use as postoperative adjuvant therapy.The breast cancer division of the Korean Society of Medical Oncology submitted an application to expand reimbursement for the postoperative adjuvant use of Roche Korea’s HER2-positive breast cancer drug, Perjeta (pertuzumab). The society has previously submitted a reimbursement application for the early-stage breast cancer indication of the CDK4/6 inhibitor ‘Verzenio (abemaciclib).’Perjeta’s adjuvant therapy indication was expected to be presented to the Cancer Disease Deliberation Committee of the Health Insurance Review and Assessment Service (HIRA) last October, but the discussion itself was canceled due to revisions to Korea’s positive listing reimbursement criteria.Consequently, it remains to be seen whether discussions regarding Perjeta’s reimbursement as postoperative adjuvant therapy will proceed this time.Currently, Perjeta is reimbursed for HER2-positive metastatic or unresectable locally recurrent breast cancer. In addition, Perjeta is reimbursed as neoadjuvant therapy in early breast cancer at a 30% patient co-insurance rate.However, postoperative adjuvant therapy, a critical treatment step to prevent recurrence, remains non-reimbursed (100% patient coinsurance rate) since the indication was added in Korea in 2018, limiting patient access.This is because its use as postoperative adjuvant therapy lacked long-term follow-up data or a high recommendation grade in global guidelines at the time of the 2019 review, unlike its use as neoadjuvant therapy (preoperative adjuvant therapy), which is covered with 30% selective reimbursement.However, the 10-year follow-up results from the global Phase III APHINITY study that was released last year are expected to fill this gap.According to the study, the Perjeta and Herceptin combination as adjuvant therapy demonstrated clear benefits, including a 21% reduction in the risk of death compared to monotherapy in patients with lymph node-positive early-stage breast cancer at high risk of recurrence.Meanwhile, the Perjeta-Herceptin combination therapy is currently recommended as Category 1 in the U.S. NCCN guidelines for postoperative adjuvant therapy in patients with HER2-positive early-stage breast cancer who have lymph node metastasis. It is also recommended as Category 1 for postoperative adjuvant therapy in high-risk patients with lymph node metastasis who achieved a pathological complete response (pCR) following neoadjuvant chemotherapy.
- Company
- 'Uplizna' to bring a shift to the NMOSD treatment strategy
- by Son, Hyung Min Apr 22, 2026 08:54am
- Tanabe Pharma Korea recently held the UPSTREAM Symposium at Voco Seoul Gangnam to celebrate the launch of NMOSD treatment Uplizna in South Korea.New treatment strategies are emerging for neuromyelitis optica spectrum disorder (NMOSD), a condition where a single relapse can lead to lifelong disability. This shift has been brought about by the introduction of 'Uplizna', a new drug targeting CD19 B cells, for NMOSD treatment in South Korea. Uplizna has been shown to significantly reduce the risk of relapse in AQP4 antibody-positive patients, and medical professionals anticipate that this treatment option will provide patients with a better choice in terms of mechanism and efficacy.Tanabe Pharma Korea recently held the UPSTREAM Symposium at Voco Seoul Gangnam to celebrate the launch of Uplizna (inebilizumab)in South Korea.Professor Ho Jin Kim of the Department of Neurology at the National Cancer Center served as the chairperson. Professor Ki Hoon Kim of the Department of Neurology at Severance Hospital and Professor Jeeyoung Oh of the Department of Neurology at Konkuk University Medical Center delivered lectures titled ▲Optimizing the treatment with inebilizumab after rituximab and ▲inebilizumab in practice: identifying appropriate patients, respectively.Uplizna is a targeted therapy for adult patients with AQP4 antibody-positive NMOSD. It works by targeting and depleting CD19+ B-cells, thereby reducing attacks (relapses) caused by the immune system targeting the optic nerve and spinal cord.The treatment received U.S. FDA approval in 2020 and was authorized in South Korea in 2021. Tanabe Pharma Korea is currently proceeding with the reimbursement listing process for the drug.During the symposium, Professor Ki Hoon Kim began by highlighting B-cells and the AQP4-IgG production pathway as core elements of NMOSD pathophysiology.The professor explained, "Plasma cells are directly linked to the clinical deterioration of NMOSD. Unlike rituximab, which targets only CD20, Uplizna depletes cells up to the pre-CD19 stage, allowing for an approach closer to the underlying cause of the disease."NMOSD is a rare autoimmune disease characterized by unpredictable, repeated relapses of optic neuritis and myelitis, leading to severe disabilities such as eye pain, blindness, and paraplegia. Approximately seven out of ten NMOSD patients are AQP4 antibody-positive; the formation of these antibodies activates the complement system, which can cause necrosis in the optic nerve and spinal cord.Notably, 80–90% of patients experience recurrent relapses, thus even a single event can result in fatal outcomes. These symptoms are often irreversible and difficult to recover from, necessitating rapid acute-phase treatment fully.Previously, NMOSD relapse relied on high-dose steroids or immunosuppressants to suppress symptoms or used rituximab or 'tocilizumab (product name: Actemra)' to prevent relapses. However, as the relapse suppression effects of these treatments were limited, there was a high demand for new options.Rituximab' clinical limitations have been pointed out. These include non-unified administration protocols, varying B-cell depletion and relapse risks based on FCGR3A genotypes, and high rates of infusion-related reactions (IRR) during the first administration. In contrast, evidence was presented that Uplizna is easy to manage with twice-yearly dosing, reduces the risk of relapse by more than 70% in clinical trials, and cuts the rate of worsening on the Expanded Disability Status Scale (EDSS) by more than half. Professor Kim summarized, "Uplizna is an option that can supplement the structural limitations for patients who do not respond to rituximab."In the second session, Professor Oh focused on the timing of switching therapies, which is a challenge in NMOSD treatment.Professor Oh emphasized, "Defining treatment failure based solely on clear relapses may cause us to miss patients." She presented various clinical patterns, including partial responses, incomplete recovery after severe relapses, and switching due to side effects or infections.Professor Oh also pointed out that due to strict domestic reimbursement criteria, some patients with "disability creep," where disability accumulates slowly without clear relapses, may miss the window for adjusting their treatment strategy.Based on actual patient cases, Professor Oh identified characteristics of patient groups requiring a switch from rituximab to Uplizna.The characteristics include ▲rapid repopulation of CD19+ B-cells, ▲incomplete response due to genotype influence, and ▲difficulty maintaining treatment due to infection burdens such as pneumonia.Professor Oh also highlighted data showing the sustained efficacy of Uplizna.The N-MOmentum study, which served as the basis for approval, was a 28-week randomized, placebo-controlled trial that faithfully reflected the NMOSD patient population in real-world clinical settings by including diverse racial and ethnic groups.In the trial, 89% of the Uplizna group remained relapse-free for 28 weeks, compared to only 58% of the placebo group.Long-term data also showed sustained efficacy, with an annualized relapse rate (ARR) of 0.03 for patients treated with Uplizna for at least 2.5 years, significantly lower than the approximately 1.0 ARR in the placebo group.However, because the open-label period (OLP) was non-blinded and uncontrolled, the results might appear more positive due to patient dropout among those with poor prognoses.Professor Oh assessed that "Uplizna is an option that is not merely as a substitute, but the one that ensures both efficacy and safety when a mechanistic switch is required."Professor Kim concluded, "Because NMOSD is a disease where a single minor relapse can lead to lifelong disability, establishing an initial strategy and determining the appropriate timing for a switch is paramount," adding, "This symposium was meaningful for refining our understanding of mechanism-based treatment and discussing customized decisions for each patient. The introduction of Uplizna will serve as an opportunity to advance NMOSD treatment strategies to the next level."

- Company
- Dupixent is shifting the atopic dermatitis treatment
- by Hwang, byoung woo Apr 22, 2026 08:53am
- Presentation by Jung Won Shin, Medical Lead at Sanofi, during the Dupixent media sessionAtopic dermatitis treatment is expanding, moving from simple symptom relief toward changing the disease progression.As treatment approaches shift toward mechanism-centered strategies following the introduction of biologics, the combination of long-term efficacy demonstrated in real-world data and early treatment strategies for children is prompting discussions of the possibility of 'disease modification.'On the 21st, Sanofi held a media session titled 'Dupixent, Rewriting the Standards of Atopic Dermatitis Treatment' to share updates on the treatment paradigm and its clinical significance.From Symptom Relief to Mechanistic Treatment…Changes in Atopic Dermatitis ApproachesAtopic dermatitis is increasingly recognized not just as a disease limited to skin symptoms, but as a chronic inflammatory condition that affects overall sleep, mental health, and comorbidities.Jung Won Shin, Medical Lead at Sanofi, explained, "Atopic dermatitis is a disease accompanied by various disease burdens in addition to visible skin symptoms," and added, "A long-term management of the disease is crucial."In particular, the emphasis was placed on the fact that the disease affects the patient's entire life, including sleep disorders, psychological withdrawal, and restrictions on social life, as well as an increased risk of infection due to skin barrier damage.These disease characteristics are leading to changes in treatment goals. In the past, the focus was on short-term symptom relief using local steroids or systemic immunosuppressants. However, the current view is that treatment strategies are moving toward targeting the underlying inflammation of the disease.Regarding this, Shin stated, "In the last 10 years, atopic dermatitis treatment has significantly changed, centering on mechanism-based targeted therapy."In this process, Dupixent was presented as a representative case leading the expansion of treatment options as a biologic that simultaneously blocks the IL-4 and IL-13 pathways.Efficacy Confirmed by Long-term Data…Strengthening Persistence·RWE EvidenceLong-term data collected in clinical settings were presented as key evidence supporting the treatment's persistence and efficacy.According to Professor Yonghyun Jang of the Department of Dermatology at Kyungpook National University Hospital, who was in charge of the presentation, an analysis of domestic patient data showed a drug persistence rate of 80.4% over 4 years.Furthermore, the EASI 75 achievement rate was 91.5%, and the EASI 90 achievement rate was approximately 44.1%, confirming that meaningful treatment responses are maintained in patients with moderate-to-severe conditions. These effects have been consistently demonstrated in actual clinical settings through global, long-term follow-up studies such as PROSE, GLOBOSTAD, and RELIEVE-AD.Professor Jang stated, "EASI, pruritus (NRS), and quality of life (DLQI) indicators all show improvement from a relatively early point after starting treatment. Significant changes in indicators appear around the three-month mark."Presentation by Professor Yonghyun Jang of the Department of Dermatology at Kyungpook National University HospitalProfessor Jang explained that these improvement effects do not end with short-term responses but tend to remain stable during long-term follow-up.Similar trends were confirmed regarding Patient-Reported Outcome (PRO) indicators.Professor Jang stated, "Meaningful improvements were demonstrated not only in objective indicators of skin lesions but also in the itching and quality of life indicators perceived by the patient. In particular, the improvement in the quality of life is characterized by being confirmed from a relatively early stage."Professor Jang added, "In the long-term follow-up data, new safety issues were limited, and there were not many cases leading to treatment discontinuation in clinical practice."Dupixent's RWE long-term follow-up data (6 years, RELIEVE-AD Registry)Attention to Early Childhood Treatment…Possibility of Disease ModificationIn particular, the media session that day presented early treatment strategies in pediatric patients and the possibility of disease modification as key points.Professor Jang said, "Atopic dermatitis is highly likely to progress along this path if inflammation is not sufficiently controlled at an early age and exacerbations are repeated," and added, "Managing the inflammatory state stably in the early stages of the disease is important to lower the long-term disease burden."Professor Jang also highlighted the role of Dupixent. This means that there is a possibility of a treatment approach that changes the natural course of the disease beyond simple symptom control.Professor Yonghyun Jang of the Department of Dermatology at Kyungpook National University HospitalAccording to the presentation, long-term follow-up studies confirmed a tendency for the disease-controlled state to be maintained after Dupixent treatment, and improvements were reported to persist in the ADCT indicator, which assesses patients' self-reported disease state.In addition, improvements continued in major symptoms, such as itching and sleep problems, which served as indicators directly linked to changes in the patient's quality of life.Furthermore, it was reported that Dupixent treatment reduced the risk of new allergies by approximately 34%, the risk of asthma by approximately 40%, and the risk of allergic rhinitis by approximately 31%.Regarding this, Professor Jang stated, "If treatment is applied early in severe patients, there is a possibility of changing the course of the disease itself in some patients. This can be seen as a treatment approach that can change long-term disease progression beyond simply controlling symptoms."During the Q&A session, it was mentioned that there are no concerns regarding long-term administration safety when administering Dupixent from childhood.Professor Jang said, "Atopic dermatitis can progress to a persistent disease once it exceeds a certain level of inflammatory state. An approach that blocks this progression through early treatment is important."Professor Jang also noted, "It is reported that the effect reappears even if Dupixent is re-administered after discontinuation. Resistance due to long-term use is not a significant concern."

- Company
- Will CDK4/6 inhibitors be reimbursed for early breast cancer?
- by Son, Hyung Min Apr 22, 2026 08:53am
- Despite significant improvements in survival rates for early-stage breast cancer patients, concerns have been raised about how treatment access to reduce long-term recurrence risk remains limited.In particular, as the clinical value of CDK4/6 inhibitors has been proven in patients at high risk of recurrence, the treatment strategy is clearly shifting beyond simple treatment toward recurrence prevention, yet it has become increasingly clear that reimbursement is failing to keep pace with these developments.On the 21st, Rep. Mi-hwa Seo of the People Power Party hosted a policy forum titled “The Era of 300,000 Female Breast Cancer Patients: Current Status and Challenges in Recurrence Management,” and intensively discussed recurrence management and treatment access issues for patients with early breast cancer.On the 21st, experts gathered at the National Assembly Members’ Office Building to discuss measures for managing recurrence in early breast cancer.Early-stage breast cancer refers to a condition where cancer cells have not spread beyond the axillary lymph nodes, and accounts for approximately 95% of all breast cancer patients.The 5-year survival rate is high, at 96.6% for Stage 1 and 91.8% for Stage 2, but the problem raised is that, regardless of survival rates, the risk of recurrence remains considerable. In particular, for hormone receptor-positive (HR+) patients, recurrence can continue beyond 5 years after diagnosis and even up to 20 years, underscoring the need for long-term management.Over the past 20 years, treatment strategies for early breast cancer have centered on surgery, radiation therapy, chemotherapy, and endocrine therapy. However, limitations regarding the inability to sufficiently reduce recurrence rates have been consistently raised.Professor Hyun-jae Yoo, Sogang UniversityIn response, a research team led by Professor Hyun-jae Yoo at Sogang University conducted a study to quantitatively analyze the socioeconomic burden on early-stage breast cancer patients.The study was conducted using a combination of surveys and expert advisory interviews with 150 patients aged 19 to 60 who had stage 1 to 3 breast cancer.The analysis revealed that patients who experienced recurrence incurred approximately KRW 29 million more in total economic losses compared to those who did not.In particular, indirect medical costs were found to increase by more than KRW 13.3 million on average, and the burden of indirect costs in recurrent patients was confirmed to be about 1.8 times higher.Professor Yoo explained, “Breast cancer is not a disease that simply ends with treatment, but a disease that affects overall quality of life and social roles after treatment. A treatment strategy that prevents recurrence has important meaning not only for the individual but also from the perspective of social cost.”Need for treatment centered on high-risk recurrence groups… “unmet need remains”Given that the risk of recurrence does not completely disappear even in early breast cancer, there is a growing consensus that more aggressive treatment strategies are needed, particularly for high-risk groups.In particular, it has been pointed out that in patient groups with a high risk of recurrence due to lymph node metastasis or tumor size, such as some late Stage 2 or Stage 3 patients, existing treatment alone has limitations.In this field, CDK4/6 inhibitor-based therapies are emerging as a new alternative. Novartis’ ‘Kisqali (ribociclib)’ and Lilly’s ‘Verzenio (abemaciclib)’ have demonstrated their efficacy in reducing the risk of recurrence through clinical trials.Professor Ji-hyun Kim, Seoul National University Bundang HospitalProfessor Ji-hyun Kim of Seoul National University Bundang Hospital emphasized, “CDK4/6 inhibitors are treatments whose efficacy in reducing recurrence and clinical utility have already been confirmed through large-scale clinical trials. They should be considered as a new standard of care for high-risk patients with early-stage breast cancer.”The problem is that these treatments are not being fully utilized in actual clinical practice.Although CDK4/6 inhibitors have been approved in Korea, reimbursement is not applied in adjuvant therapy for early breast cancer, forcing patients to bear the full cost of these expensive drugs.In contrast, Verzenio is already covered by national health insurance in major countries such as the United Kingdom, Australia, Canada, France, Germany, Italy, China, and Singapore, ensuring access to treatment.Professor Ji-hyun Kim added, “From the perspective of an individual patient, one would want to try every possible treatment, but in terms of national finances, there is the difficulty of balancing resources across various diseases. There needs to be a social discussion on how to rate the value of therapies that prevent recurrence in advance.”“Financial resources and equity are variables”… attention on government’s judgment over reimbursement criteriaIn the panel discussion that followed, policy issues surrounding access to new drugs for early breast cancer were intensively discussed.Professor Seong-bae Kim of Asan Medical Center in Seoul, who moderated the discussion, emphasized, “While triple-negative breast cancer is generally known to be more aggressive, some hormone receptor-positive breast cancers also present a higher risk of recurrence. It is important to identify high-risk groups and apply appropriate treatments.”He added, “The efficacy of CDK4/6 inhibitors has already been clinically proven. The issue lies in reaching a social consensus on how to apply this in actual clinical practice and which patient groups should be prioritized within limited financial resources.”Professor In-Hae Park of Korea University Guro Hospital said, “Even in high-risk patients, there are cases in which it is difficult to actively explain or recommend treatment because of the economic burden. The reality itself, in which treatment strategy must be adjusted in consideration of the patient’s financial situation, places a burden on medical staff as well.”The panelists also pointed out that the threshold for reimbursement has risen as the weight of financial impact assessments has increased in recent Cancer Disease Deliberation Committee meetings.A view of the panel discussionYoon-ho Eo, a reporter for Daily Pharm, stated, “Currently, the results of reimbursement reviews are structured such that only the decision to approve or deny coverage is disclosed, lacking transparency regarding the basis for the judgment. There is a need to strengthen the transparency and flexibility of the evaluation process.”The government maintains a cautious stance but intends to continue related discussions.Min-jung Kim, an official at the Division of Pharmaceutical Benefits at the Ministry of Health and Welfare, stated, “Reimbursement applications for Verzenio and Kisqali have been submitted, and discussions are expected to proceed in the Cancer Disease Deliberation Committee within the first half of the year. We plan to conduct a fair review by comprehensively considering clinical utility, patient access, and the impact on National Health Insurance finances.”She continued, “In line with the recent trend toward strengthening coverage for anticancer drugs, the scope of reimbursement has been continuously expanding. Since equity must be maintained within limited financial resources, careful review of the patient group setting and the scope of application is necessary.”The Health Insurance Review and Assessment Service presented a similar position.Ae-ran Park, Director of the Pharmaceutical Benefits Standards Division at HIRA, explained, “We are fully aware of the clinical necessity and field demands for preventing early recurrence of breast cancer. The demand to apply follow-up treatments at an early stage is a trend common not only in breast cancer but across various cancer types.”Park added, “While expanding treatment accessibility based on clinical utility is important, we must also consider the balance with the sustainability of the National Health Insurance finances. Setting the criteria for which patient groups should be prioritized is the key task.”

- Company
- Merz Aesthetics Korea successfully holds ‘Confidence To Be Conference’
- by Hwang, byoung woo Apr 21, 2026 07:33am
- Scene from the ‘Confidence To Be Conference 2026’ eventMerz Aesthetics Korea announced on the 20th that it held the “Confidence To Be Conference 2026.”The event was held on the 16th at Dongdaemun Design Plaza in Seoul under the theme “Finding the answer in being myself,” and was planned as part of the company’s global consumer ESG campaign “Confidence To Be.”The event was also based on the company’s mission, “Look Better, Feel Better, Live Better,” helping all people live with greater confidence through the journey of finding a better self.The event highlighted the correlation between medical aesthetics and confidence, drawing on data from the global study “Pillars of Confidence,” which surveyed 15,000 people across 15 countries, as well as big-data trends.Through this, the company presented expanded value that extends beyond individual physical transformation to include enhanced social confidence and improved quality of life.Experts from various fields then participated to share their experiences and philosophies of living a confident life.Strength coach Eunseo Kim emphasized the importance of self-understanding and attitude and conducted practical exercises on site, while Mickey Kim introduced his experience of career transition and mindset for change. Choreographer Monica highlighted the importance of making choices at life’s turning points and living proactively.In addition, the event addressed the importance of proper self-perception in the social media environment and presented a healthy approach to medical aesthetics, including consultations with medical professionals, long-term planning, and the sharing of procedure history.Su Yeon Yu, General Manager of Merz Aesthetics Korea, said, “Medical aesthetics is not about standardized beauty, but a process of enhancing quality of life through confidence that is true to oneself. We will continue to expand our positive impact.”

- Company
- "Companion diagnostics…ovarian cancer diagnostic paradigm shift"
- by Hwang, byoung woo Apr 21, 2026 07:33am
- A shift in the treatment paradigm for platinum-resistant ovarian cancer, a field with historically limited options, is anticipated following the arrival of antibody-drug conjugate (ADC) therapies.Notably, the introduction of new treatments has highlighted the growing importance of companion diagnostics in determining 'which patients should receive the therapy?'Daily Pharm met with Min Ae Baek, Manager of the Pathology Diagnostics Division at Roche Diagnostics Korea, to discuss the significance of companion diagnostics in ovarian cancer, practical clinical application, and the substantial value to patients.Platinum-resistant ovarian cancer, a disease without alternative options…addressing treatments and diagnostics altogetherOvarian cancer is classified as a high-risk malignancy due to the difficulty of early detection and high recurrence rates. Most patients are diagnosed at advanced stages and frequently experience recurrence even after standard therapy. Platinum-resistant ovarian cancer, which recurs within six months of platinum-based chemotherapy, poses a particularly restrictive environment due to low response rates to conventional therapies and poor prognosis.Manager of the Pathology Diagnostics Division at Roche Diagnostics KoreaBaek identified the 'treatment void' as the primary unmet need in platinum-resistant ovarian cancer. Baek explained, "Reaching the platinum-resistant stage effectively leaves patients with no viable alternatives. As response rates to existing drugs decrease and life expectancy drops to approximately one year, both patients and guardians find themselves in a desperate situation," she explained.Baek said, "Most patients with ovarian cancer are diagnosed at stage 3-4, and in many cases, patients experience relapses; the majority of patients reach these stages."The recently introduced targeted therapy, Elahere (mirvetuximab soravtansine), and its companion diagnostic are viewed as pivotal elements that could alter the current treatment landscape.Baek said, "The emergence of a new option in an area where alternatives were non-existent, coupled with a test to screen for patients who can benefit from it, represents a significant evolution."Treatment success is dependent on patient selection…the significance of companion diagnosticsThe oncology paradigm is rapidly evolving. Previously, treatment relied on broad application. Currently, it is moving toward precision medicine based on specific biomarkers. In this transition, companion diagnostics function as the starting point of the treatment strategy."The core value of companion diagnostics lies in selecting the appropriate patient group, thereby simultaneously enhancing treatment efficiency and safety," Baek stated. "Identifying suitable patients through testing reduces the burden of side effects and costs associated with unnecessary treatments while increasing the efficiency of the clinical decision-making process."Baek's view is that these trajectories not only help medical professionals to make decisions but also directly impact patients.At the center of this shift is the biomarker folate receptor alpha (FRα). As a protein highly expressed on ovarian cancer cells, FRα has gained clinical utility as a target for ADC therapies. Baek explained, "Ovarian cancers require folate for proliferation and overexpress these receptors to absorb folate for proliferation. This characteristic is utilized for targeted anticancer strategies."The VENTANA FOLR1 (FOLR1-2.1) RxDx Assay plays a crucial role here. This immunohistochemistry (IHC)-based companion diagnostic assesses FRα expression in tumor tissue to identify eligible patients. Baek mentioned, "This approach is a dye-based assay, where FRα protein expression is checked, and pathologists confirm these under a microscope," and "It does not simply check for presence but evaluates expression levels against criteria to determine if treatment is applicable."The value of diagnostic-led therapy has been substantiated in clinical studies such as the Phase 2 SORAYA and Phase 3 MIRASOL trials. Baek stated, "The SORAYA study provided initial evidence showing a meaningful objective response rate for Elahere monotherapy," and added, "In the MIRASOL study, superior therapeutic effects compared to conventional chemotherapy were confirmed when treatment was applied after selecting patients with high FRα expression.Specifically, in clinical trials, Elahere has been shown to improve major clinical indicators compared to existing treatments. Significant improvements in progression-free survival and objective response rate have been reported, along with a reduction in the risk of disease progression or death.Current guidelines from the Korean Society of Gynecologic Oncology (KSGO) already recommend Elahere for FRα-positive platinum-resistant ovarian cancer with the highest level of evidence (Level I) and recommendation grade (Grade A).Domestic launch imminent in May..."Will help in deciding treatment options"In Korea, the treatment environment based on companion diagnostics is also entering the full-scale introduction stage.Both Elahere and Roche Diagnostics Korea's companion diagnostic (VENTANA FOLR1 (FOLR1-2.1) RxDx Assay) have already received approval from the Ministry of Food and Drug Safety, and clinical use is expected once test supplies begin.Baek said, "To prescribe medicines, assays must be available. Assays will be supplied in Korea starting this May," and added, "Pathological divisions in Korea already have an established platform to deliver these assays. The view is that it will be effectively implemented, considering the established diagnostics infrastructure."Baek also mentioned, "There have been continuous inquiries from the field regarding the ability to provide new treatment options to patients with platinum-resistant ovarian cancer," and conveyed, "As it is a test that already has high awareness among medical staff and has been awaited for introduction, expectations are also high from patients."As the treatment and companion diagnostic are ahead of their launch, there is an opinion in the clinical field that it is necessary to preemptively establish treatment plans by performing the FRα test during the initial diagnosis stage rather than at the stage of treatment use.Regarding this, Baek projected that if the test is conducted within the approved criteria, it could help consider future treatment options.Baek stated, "The current approval criteria allow for testing on specimens from patients diagnosed with high-grade serous epithelial ovarian cancer, fallopian tube cancer, or primary peritoneal cancer," and noted, "Checking each patient's information in advance could assist medical staff in establishing treatment plans."Ultimately, as the expansion of companion diagnostics leads beyond simple technology introduction to a change in the treatment structure, the role of Roche Diagnostics Korea is also expanding.The company is focusing on more than just supplying testing products. It is building an environment where pathology medical staff can perform tests reliably and support the smooth implementation of companion diagnostic-based treatment.Baek added, "As companion diagnostics are a critical area directly linked to treatment, it is important to ensure that patients are appropriately selected through standardized tests in a stable environment and that the results lead to treatment," and added, "We will continue to prepare the foundation so that companion diagnostics can be implemented in the medical field in line with the trend of emerging new biomarkers and treatments."
- Company
- ‘Lower and Faster’…Dyslipidemia treatment strategy evolves
- by Son, Hyung Min Apr 21, 2026 07:33am
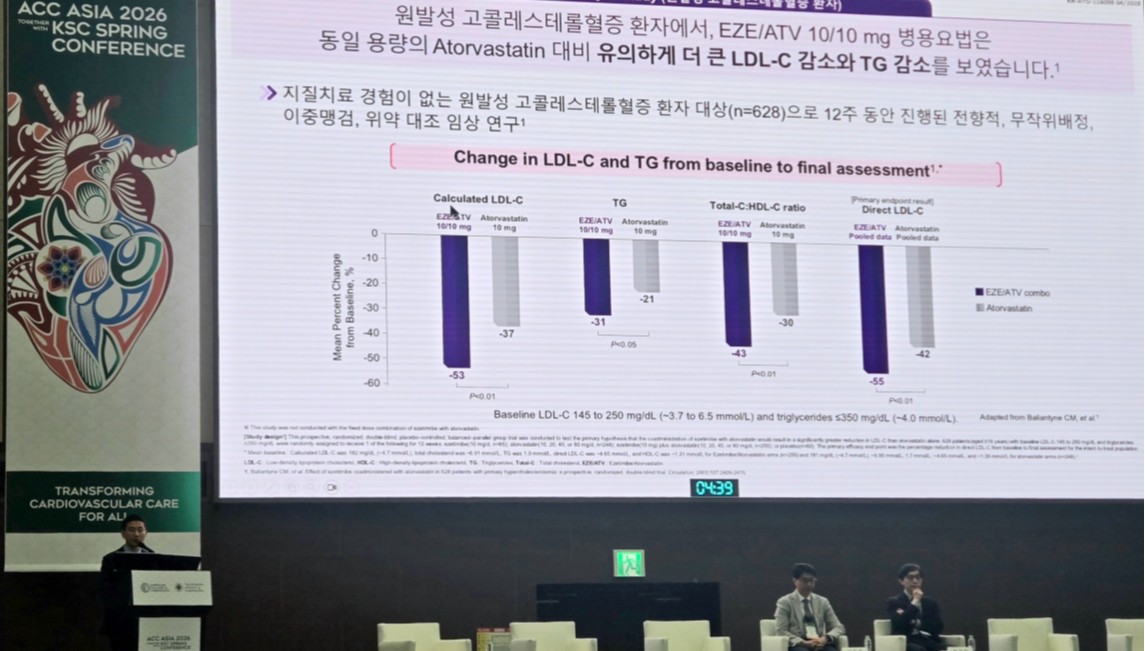
- Beyond the question of “how low to lower” LDL cholesterol, “how quickly to reach the target level” is emerging as a key variable in the treatment of dyslipidemia.As the target has been tightened to below 55 mg/dL, an approach aimed at reaching the goal quickly through initial combination therapy is emerging as an alternative.According to industry sources on the 21st, treatment strategies for dyslipidemia based on early combination therapy were presented as a major topic at the ACC Asia 2026 Together With KSC Spring Conference held recently in Gyeongju.Cardiovascular disease is one of the major causes of death in Korea, and dyslipidemia is considered a representative risk factor that can be corrected through treatment.Nevertheless, the 30-day and 1-year mortality rates of acute myocardial infarction patients in Korea still stand at approximately 9% and 16%, respectively, indicating that existing treatment strategies alone have not been sufficient to adequately reduce early mortality risk. This, together with the fact that the LDL cholesterol target attainment rate in very high-risk patients remains limited, suggests the need for a more aggressive treatment approach.Against this backdrop, the need for a combination therapy that can overcome the limitations of statin monotherapy has been consistently raised. Notably, the IMPROVE-IT study reported that adding ezetimibe to statins resulted in an additional approximately 24% reduction in LDL-cholesterol, along with a 24% and 32% reduction in the risk of myocardial infarction and ischemic stroke, respectively.With long-term safety and efficacy subsequently confirmed, ezetimibe has become established in major guidelines as a preferred combination and second-line treatment option.Professor Jin Wi of the Division of Cardiology at Gachon University Gil Medical Center emphasized the importance of statin + ezetimibe combination therapy at the ACC Asia 2026 Together With KSC Spring Conference.Professor Jin Wi of the Division of Cardiology at Gachon University Gil Medical Center said, “Dyslipidemia treatment is shifting from a high-intensity statin-centered approach to a more aggressive cholesterol-lowering strategy. It is important to reach LDL cholesterol targets quickly through early combination therapy.”In particular, he emphasized the importance of dyslipidemia management from a CKM (Cardio-Kidney-Metabolic) perspective, where cardiovascular, kidney, and metabolic diseases are interconnected, saying, “In these patients especially, LDL cholesterol should be controlled aggressively at an early stage.”He added, “Atorvastatin can be used in chronic kidney disease patients without dose adjustment, which gives it high clinical utility.”Professor Suk Min Seo, Division of Cardiology at Eunpyeong St. Mary’s Hospital, also noted, “In actual clinical practice, it is difficult to fully implement stepwise therapy due to limitations in patient follow-up. Applying high-intensity statin and ezetimibe combination therapy from the outset is a realistic strategy in terms of achieving the target within a short period and managing risk.”At the session that day, the ezetimibe/atorvastatin combination product ‘Atozet’ was also introduced as one of the initial combination therapy options.Beyond ‘lower’ to ‘faster’… guidelines and clinical evidence alignRecent guidelines are demanding changes not only in treatment goals but also in the treatment approach itself.The 2025 European Society of Cardiology / European Atherosclerosis Society (ESC/EAS) guidelines presented the LDL cholesterol goal for patients with atherosclerotic cardiovascular disease (ASCVD) as below 55 mg/dL and at least a 50% reduction from baseline, and in some very high-risk groups, recommended below 40 mg/dL.Furthermore, for treatment-naïve ACS patients, the guidelines explicitly recommend considering combination therapy with high-intensity statins and ezetimibe from the outset, emphasizing aggressive intervention at the start of treatment.The 2026 American College of Cardiology / American Heart Association (ACC/AHA) guidelines likewise presented an LDL cholesterol goal of below 55 mg/dL for very high-risk patients in secondary prevention, and recommend early addition of non-statin agents when the target is difficult to achieve with statin monotherapy alone. Accordingly, the treatment paradigm appears to be shifting from the existing stepwise approach to an early combination-centered strategy.Professor Suk Min Seo, Division of Cardiology at Eunpyeong St. Mary’s Hospital, presenting at the ACC Asia 2026 Together With KSC Spring ConferenceThis shift is supported by clinical research. The Ez-PAVE trial was a study designed to verify the clinical validity of lowering the LDL cholesterol target from the conventional 70 mg/dL to 55 mg/dL.In this study, which enrolled 3,048 Korean ASCVD patients, the group targeting an LDL-cholesterol level below 55 mg/dL achieved a statistically significant reduction of approximately 33% in major adverse cardiovascular events (MACE) over three years compared to the group targeting below 70 mg/dL. This suggests that a strategy of controlling LDL cholesterol to a lower level can lead not only to numerical improvement but also to an actual reduction in clinical events.Furthermore, the BETTER TRIAL study showed that early combination therapy resulted in significant improvements in both the magnitude of LDL-cholesterol reduction and the rate of target achievement compared to monotherapy. In this study of Korean patients with very high-risk ASCVD, early combination therapy with ezetimibe and atorvastatin, administered before reaching the maximum statin dose, demonstrated significant improvements in both the magnitude of LDL-cholesterol reduction and the rate of target achievement compared to monotherapy.In particular, the rate of achieving LDL-cholesterol levels below 55 mg/dL was 46.2% versus 9.0% at 6 weeks and 55.0% versus 15.4% at 12 weeks, clearly demonstrating the efficacy of the early combination strategy.Ultimately, the treatment of dyslipidemia is expanding beyond the question of “how low to lower” to include “how quickly to achieve” the target level. Considering changes in guidelines and clinical evidence, a strategy of applying aggressive combination therapy from the outset is expected to emerge as a key factor in improving patient outcomes.

- Company
- Amgen’s BiTE platform to become a pillar of solid tumor strategy
- by Son, Hyung Min Apr 20, 2026 04:58pm
- Bispecific antibody-based immuno-oncology platforms are expanding beyond hematologic malignancies into solid tumors, emerging as a new axis of treatment strategy transformation.Amgen is rapidly expanding its position as a next-generation immuno-oncology strategy by broadening the application scope of its ‘BiTE (Bispecific T-cell Engager)’ technology based on a research and development system that combines genetics and artificial intelligence (AI).Meejin Cho, Hematology-Oncology TA Lead, Medical Affairs, Amgen KoreaOn the 17th, Amgen Korea held a media session at its Seoul headquarters under the theme “AMGEN INNOVATION TALK BiTE – Innovation Driving Changes in Patient Treatment,” and unveiled its R&D strategy and direction for BiTE platform expansion.Under its mission of “To Serve Patients,” Amgen has been developing treatment options in areas of serious diseases such as cancer, cardiovascular disease, inflammation, and rare diseases. Recently, the company has been focusing on simultaneously enhancing the precision and speed of new drug development by integrating AI technology based on its understanding of human genetics and disease biology.The R&D strategy is structured around three pillars: ▲identifying the root causes of diseases and discovering new targets, ▲developing diverse treatment modalities, and ▲innovating clinical trial design.These strategies are being implemented in actual research processes. Amgen uses large-scale genetic data accumulated through its subsidiary deCODE Genetics to analyze disease causes and treatment responses with high precision. In addition, it applies the AI platform ‘Freyja,’ developed in collaboration with NVIDIA, to predict the probability of success of candidate substances in advance, thereby improving development efficiency.The use of AI is also expanding in the clinical stage. By analyzing real-world patient data through its proprietary machine learning model ‘ATOMIC’ and identifying institutions with a high likelihood of participating in clinical trials, the company claims to have increased patient recruitment speed by approximately threefold compared to previous methods. This directly contributes to shortening the drug development timeline and improving patient access.On this foundation, Amgen’s core platform is BiTE. BiTE is a bispecific antibody-based immuno-oncology platform that enables a patient’s immune T cells to directly recognize and attack cancer cells.Cancer cells evade immune surveillance through various mechanisms. While existing immune checkpoint inhibitors restore T-cell activity by blocking the PD-1/PD-L1 binding, BiTE represents a more advanced approach.In some cancers, cells evade T-cell recognition by reducing MHC class I expression, but BiTE bypasses this limitation. Its structure allows it to bind simultaneously to the T-cell’s CD3 and the target antigen on the cancer cell, thereby inducing immune cells to directly attack cancer cells without relying on the TCR-MHC class I recognition process.In other words, unlike previous approaches that only activated immune responses, BiTE physically connects T cells and cancer cells, inducing immediate cell death.Based on these mechanistic strengths, BiTE is expanding its application across various cancer types and has already proven its clinical value in hematologic malignancies.‘Blincyto (blinatumomab),’ a treatment for acute lymphoblastic leukemia (ALL), induces anti-cancer effects by linking CD19-positive B cells and T cells and has demonstrated improved survival compared to existing treatments. Based on this evidence, it is recommended as first-line consolidation therapy in the NCCN guidelines.Building on this success, Amgen is expanding the application of BiTE technology to solid tumors. A prime example is “Imdelltra (tarlatamab),” a treatment for extensive-stage small cell lung cancer.Small-cell lung cancer is a disease with rapid progression and high recurrence rates, representing an area of significant unmet medical need. Imdelltra is a BiTE-based therapy that targets DLL3 to induce T cells to directly attack cancer cells.Clinical results have also demonstrated meaningful outcomes. In patients whose disease progressed after receiving at least two prior treatments, including platinum-based chemotherapy, Imdelltra achieved an objective response rate of approximately 40% and a median overall survival (mOS) of 14.3 months. In the Asian patient population, this was extended to 19 months.Based on these data, Imdelltra was approved in the United States in 2024 and recommended as a subsequent therapy (Category 1) in NCCN guidelines. In Korea, it has been designated as a Global Innovative product on Fast Track (GIFT) and approved, with reimbursement procedures currently underway.The emergence of Imdelltra is evaluated as a turning point in small cell lung cancer treatment, which has seen little change over the past 30 years.Meejin Cho, Hematology-Oncology TA Lead at Amgen Korea, explained, “BiTE is particularly effective in diseases with clear target antigens and rapid progression. Expanding from ALL to small cell lung cancer, which has a clear target, DLL3, is an extension of this strategy.”She added, “Among our current pipeline, we view prostate cancer as the area closest to commercialization. We will continue to expand our scope of application, focusing on cancer types with high unmet medical needs.”

- Company
- GE HealthCare rebounded with 300B won sales last year
- by Hwang, byoung woo Apr 20, 2026 04:58pm
- GE HealthCare, which is pushing for an "Integrated Precision Healthcare" that combines hardware with digital solutions, successfully led a sales rebound last year.Following the medical-government conflict that hit the industry in 2024, the expansion of communication with hospitals and clinics in South Korea via digital platforms is cited as a key factor in improving performance.After the 2024 medical-government crisis…V-shaped rebound in one year According to recently released audit reports, GE HealthCare Korea's sales in 2025 reached KRW 317.1 billion, a 14.9% increase from KRW 275.8 billion the previous year.After sales dropped 6.8% in 2024 to KRW 295.9 billion from 2023, the company quickly surpassed the KRW 300 billion the following year, signaling a significant recovery.Operating profit also increased by 13.4%, from KRW 16.86 billion in 2024 to KRW 19.13 billion in 2025. During the same period, net income rose from KRW 14.57 billion to KRW 17.09 billion. However, the operating profit margin remained stable at around 6.0% (6.1% in 2023, 6.1% in 2024, and 6.0% in 2025), indicating steady maintenance rather than a drastic improvement in profitability.GE HealthCare Korea's 4-Year Sales & Operating Profits: GE HealthCare Korea's sales in 2025 reached KRW 317.1 billion. Source: FSS DART, unit: KRW 100 million (Audit report re-graphed by Daily Pharm)As the medical-government conflict issues gradually resolve, the company's efforts to strengthen capabilities and improve its internal structure have been proven through these figures, suggesting a structural rebound rather than a simple recovery.In 2024, the decline in sales was largely due to a decrease in product sale, which dropped from KRW 193.8 billion to KRW 170.8 billion.In contrast, service sales increased from KRW 102.1 billion to KRW 104.9 billion during the same period, acting as a buffer while equipment sales were shaken.In 2025, both products and services rebounded simultaneously. Product sales reached KRW 203.9 billion, and service sales rose to KRW 113.2 billion.GE HealthCare Korea's Product Sales & Service Sales Ratio: GREEN-product sales, BLUE-service sales. Source: FSS DART, Unit: KRW 100 million. (Audit report re-graphed by Daily Pharm)GE HealthCare strengthens platform strategy…Expanding hospital collaborationsGE HealthCare is shifting its weight from a structure centered on equipment sales to a platform-based strategy.The core of this is the 'D3 Strategy,' which encompasses Device, Disease, and Digital. It aims to use AI to improve quality, support customized treatment, and ultimately integrate data into a cloud-based platform.Globally, GE HealthCare has obtained FDA clearance for 100 AI-enabled medical devices, setting a record for the most listings in four consecutive years.A representative technology, 'AIR Recon DL,' which offers both reduced MRI scan times and improved image quality, has been introduced to more than 20 medical institutions in Korea.SPI index displayed on the CARESCAPE CanvasAdditionally, in February, the Surgical Pleth Index (SPI), a monitoring indicator that quantifies pain responses during surgery, was listed as a "New Medical Technology." This expanded the company's presence beyond imaging into the operating room and patient monitoring sectors.The technology portfolio is shifting from simple imaging equipment to clinical decision-support tools.Consequently, partnerships with major hospitals are expanding beyond simple supply contracts into a system of "Reference Sites" and cooperative hospital networks.For example, Dongtan City Hospital was designated as an Asian regional reference site in January. Its AI Radiology Center serves as a hub where customers from Korea, ASEAN, Australia, and New Zealand can refer to operational cases.Gumi Gangdong Hospital signed an MOU for clinical cooperation and equipment operational efficiency. Daegu Miracle Women's Clinic is partnering to advance infertility treatment based on high-end ultrasound technology.These structures integrates everything from equipment supply to operational training, protocol sharing, and the expansion of clinical utilization into a single package.Yong-duck Kim, President & CEO of GE HealthCare Korea, stated, "The designation of Dongtan City Hospital as an Asian regional reference site signifies that the equipment operation and clinical environment of medical institutions in South Korea have been recognized as excellent global reference cases," and added, "GE HealthCare will continue to actively support Dongtan City Hospital in maintaining efficient and stable equipment operation and building a sustainable medical environment."