- LOGIN
- MemberShip
- 2026-06-21 03:57:17

- Company
- ‘Bispecific antibodies gain attention as option for DLBCL'
- by Whang, byung-woo May 21, 2025 06:36am
- Diffuse large B-cell lymphoma (DLBCL) is considered a difficult disease to treat due to its aggressive nature, but the emergence of new drugs has brought about a paradigm shift in its treatment. As efforts for DLBCL progress towards a cure beyond extending survival, the role of bispecific antibody therapies is garnering attention. Georg Lenz, Professor at Münster University Hospital in Germany, and Jung-ok Lee, Professor of Hematology and Oncology at Seoul National University Bundang Hospital, emphasized the diversified DLBCL treatment options and the importance of timely treatment for maximizing treatment efficacy during their interview with Daily Pharm. Professor Georg Lenz (Münster University Hospital) Approximately 60% of DLBCL patients can expect a cure with first-line treatment, but around 30–40% of the remaining patients are resistant to first-line treatment or experience relapse. Although no two patients are in the same situation, the two professors explained that achieving complete remission remains the primary goal, even for patients with relapsed or refractory (r/r) disease or those who have undergone third-line or later-line treatments. Innovative new drugs such as “bispecific antibodies” and “antibody-drug conjugates (ADC)” are playing a major role in this process. However, DLBCL, which is characterized by rapid progression and aggressive characteristics among non-Hodgkin's lymphomas, is one disease that is difficult to cure. Overseas, particularly in countries such as Germany, CAR-T therapy and bispecific antibody therapies have become clear treatment options depending on the recurrence period and health status of DLBCL patients. According to Professor Georg, Germany uses various bispecific antibody therapies, including Epkinly, glofitamab, and odronextamab in the third-line treatment stage. Professor Georg explained, “For r/r DLBCL patients receiving third-line treatment, bispecific antibody therapies become a treatment option. If CAR-T therapy was not administered in previous treatment stages, CAR-T therapy can also be considered as a treatment option in the third-line treatment stage.” In contrast, in South Korea, the only CAR-T therapy available after second-line treatment is Kymriah, and bispecific antibody therapies are not reimbursed. Regarding this, Professor Lee stated, “Polatuzumab vedotin in combination with R-CHP improved progression-free survival (PFS) compared to R-CHOP in the first-line treatment of DLBCL, and it is being used as a first-line treatment in several countries overseas. However, in Korea, R-CHOP remains the standard of care, so the rate of recurrence after first-line treatment will be relatively high.” In particular, the difference in treatment access between Korea and other countries is evident after second-line treatment. This is because while three CAR-T therapies have been approved in the United States, only one, Kymriah, has been approved in Korea. Professor Lee emphasized, “The domestic treatment environment falls short of global standards, and patients are missing out on real treatment opportunities. In addition to Kymriah, it is urgent to introduce additional CAR-T therapies such as Axi-cel and Liso-cel and apply reimbursement to bispecific antibodies such as Epkinly.” With Epkinly prescriptions on the rise, the challenge is to overcome the remaining “treatment hurdles” The reason why it is important for new drugs such as Epkinly, a bispecific antibody, to enter the regulatory system is because the ultimate goal of r/r DLBCL treatment is a complete cure. Although bispecific antibodies lack long-term data compared to CAR-T therapies, which were developed earlier, they are considered to be a useful treatment option due to their relatively lower toxicity for elderly patients and others who cannot receive CAR-T therapy. Professor Jeong-Ok Lee (Seoul National University Bundang Hospital) Professor Georg said, “I believe that approximately 3 years of long-term data accumulation will be sufficient. Considering the rapid changes in the treatment environment and the various data, I think there is a high possibility that Epkinly will firmly establish itself in the second-line treatment area in the near future.” He added, “Based on the clinical data disclosed to date, the results of Epkinly combination therapy are showing positive trends. We optimistically anticipate that Epkinly may advance into an earlier line treatment option or even become a first-line treatment option used before the r/r stage, rather than completely replacing CAR-T therapy.” According to three-year follow-up data presented at the American Society of Hematology (ASH) 2024 Annual Meeting, Epkinly achieved an overall response rate (ORR) of 59%, with 41% achieving complete remission. Professor Lee explained, “It is encouraging that the 36-month survival rate of patients that reached complete remission was 63%. Additionally, the fact that the progression-free survival (PFS) curve showed a stable ‘plateau’ pattern after a certain point is a positive signal that the treatment effect may persist in the long term.” Although direct comparison is difficult as long-term follow-up data has not yet been accumulated as much as for CAR-T therapy, Professor Lee believes that Epkinly is also expected to achieve high response rates, complete remission, and sustained response. Clear differences exist in the global and domestic treatment environments… reimbursement needs to be realized There are various conflicting opinions regarding the current position of CAR-T therapy and bispecific antibody therapy, including their complementary positions as a 'bridge therapy' before CAR-T therapy. Both professors stated that it is difficult to determine the exact order considering reimbursement conditions and their clinical experience, but mentioned that Epkinly may be considered over CAR-T therapy in elderly patients in terms of adverse reaction management. Professor Georg explained, “Epkinly has shown meaningful clinical efficacy in elderly or refractory patients who are difficult to treat with CAR-T therapy. In particular, it has a low and predictable incidence of cytokine release syndrome (CRS) and has been reported to be associated with minimal neurotoxicity in the form of Immune Effector Cell-Associated Neurotoxicity Syndrome (ICANS), making it highly safe.” He added, “CAR-T therapy takes about a month to manufacture, so Epkinly may be considered first when time is limited. Although there are no age restrictions for either Epkinly or CAR-T, Epkinly may be a more appropriate choice for patients who are in poor overall health or are elderly.” The problem remains that the reimbursement situation in Korea is still lacking in terms of a global standard of care. He emphasized the need to improve the reimbursement system and access to treatment so that DLBCL patients in Korea can receive the latest global standard treatment. Professor Lee said, “In South Korea, there is a gap between the global standard and Korea’s treatment in all lines of treatment, from the first to the third. Therefore, other effective CAR-T therapies besides Kymriah should be introduced as soon as possible, and bispecific antibody therapies, including Epkinly, should be included in the health insurance reimbursement system.” Finally, he added, “As a hematologist treating DLBCL patients and a member of the Korean Society of Hematology, I hope that a medical environment is established where patients can receive bispecific antibody therapy as soon as possible. I feel a sense of responsibility as an expert and plan to actively voice my opinions and fulfill my role in both my academic society and as an individual physician.”
- Policy
- 'AI-enabled stem cell for pediatric epilepsy proves effect'
- by Lee, Hye-Kyung May 21, 2025 06:36am
- A new treatment possibility has opened up for pediatric epilepsy patients who have shown little response to existing treatments. The Korea Health Industry Development Institute (President: Soondo Cha) announced on the 19th that Professor Hoon-Chul Kang’s research team at Yonsei University Severance Children's Hospital has successfully discovered a new drug candidate personalized for patients and experimentally proven its efficacy by utilizing AI-based drug discovery technology and induced pluripotent stem cells (iPSCs) obtained from patients. Pediatric epilepsy is a representative intractable neurological disorder, affecting approximately 250,000 people in Korea, with about 30-40% of patients suffering from drug-resistant epilepsy that does not respond to existing antiepileptic drugs. (from the left) Professor Hoon-Chul Kang and Ji-hoon Kim from Yonsei University and Professor Do Kyun Na from Chung Ang University In particular, pediatric epilepsy patients with rare genetic mutations, such as SCN2A gene mutations, exhibit significant differences in experimental responses by individual patient, rendering it difficult to achieve significant improvements with existing treatments. In addition, the lack of precise disease models and appropriate drug screening technologies has made the development of personalized drugs an urgent necessity. A joint research team led by Professor Hoon-Chul Kang and Ji-hoon Kim from Yonsei University and Professor Do Kyun Na from Chung Ang University addressed this issue by creating induced pluripotent stem cells (iPSCs) from patients' blood cells to develop precision disease models in the same disease environment as actual patients. Also, the team successfully identified new drug candidates, surpassing existing treatments through AI-driven high-throughput compound screening and validation. The research team used the latest gene editing technology to correct the SCN2A mutation to its normal state to reveal that the mutation is the direct cause of epilepsy, and confirmed that seizure symptoms disappeared. Furthermore, based on personalized neural cell models, the team analyzed approximately 1.6 million compounds using AI-based simulations and selected 5 optimal new drug candidates by considering blood-brain barrier permeability, toxicity, and gene binding affinity. Among these, 2 candidates demonstrated approximately 100 times higher efficacy than the existing treatment phenytoin, paving the way for personalized treatment. This study can be evaluated as a case of precision medicine that provides a practical alternative for intractable patients whose symptoms have not improved with existing treatments by combining the genetic characteristics of patients with rare genetic diseases with cell-based models and AI-based new drug discovery techniques. Professor Kang explained, “This study is a case where patient-derived cell-based personalized drug discovery technology for patients with intractable epilepsy demonstrated an effect. We plan to continue expanding our research to develop personalized precision therapies for patients with various genetic mutations, including SCN2A.” Professor Na emphasized, “This study demonstrates the practical applicability of precision medicine technology in the field of rare diseases. We believe that the findings from this study will help establish an innovative treatment strategy that can be applied to patients with various genetic disorders in the future.” This study was conducted with support from the Public Health Technology Research Project promoted by the Ministry of Health and Welfare and the Korea Health Industry Development Institute, and was published in the 2025 issue of Computers in Biology and Medicine, a world-renowned academic journal in the field of medical information.
- Company
- Trodelvy may be the 1st drug reimbursed with ICER benefits
- by Eo, Yun-Ho May 21, 2025 06:36am
- ADC breast cancer drug Trodelvy may soon be listed for insurance reimbursement in Korea. Gilead Sciences recently finalized price negotiations with the National Health Insurance Service for its triple-negative breast cancer (TNBC) treatment Trodelvy (sacituzumab govitecan). As a result, Trodelvy is scheduled to be presented at the Health Insurance Policy Deliberation Committee meeting this month, and if approved, the drug could be listed as early as June. Last August, the detailed evaluation criteria for new drugs subject to price negotiations were revised, and Trodelvy has met the criteria for innovative new drugs, marking the first case where the incremental cost-effectiveness ratio (ICER) threshold was applied flexibly for reimbursement in Korea. While there have been previous cases where an exceptional ICER threshold was applied to an antibody-drug conjugate (ADC) like Trodelvy, such as “Enhertu (trastuzumab deruxtecan),” the fact that Trodelvy is the first drug to be subject to the revised criteria after the revised guidelines were implemented is significant. Although there are no clearly documented figures, the generally accepted maximum ICER threshold for insurance reimbursement in Korea is KRW 50 million. Even cases where the KRW 50 million threshold was approved are said to be extremely rare. Trodelvy's threshold is known to exceed KRW 70 million. With the first tape cut, it will be worth watching how many more drugs will qualify for such ICER benefits following Trodelvy. Triple-negative breast cancer is an aggressive form of breast cancer that recurs and metastasizes rapidly. Patients with metastatic triple-negative breast cancer who have metastasized despite treatment have a life expectancy of only a few months, even with chemotherapy. However, chemotherapy has long been the standard of care due to the lack of targets that can effectively kill cancer cells. Trodelvy, the first Trop-2-targeted antibody-drug conjugate (ADC), is the only treatment for metastatic triple-negative breast cancer in the second-line or higher setting that has been shown to prolong survival compared to chemotherapy and has settled as the global standard of care since its introduction. Currently, major guidelines in the U.S. and Europe specify Trodelvy as the preferred agent for patients with previously treated metastatic triple-negative breast cancer. In a Phase III study, the overall survival of the chemotherapy arm was 6.9 months, compared to a nearly one-year survival (11.8 months) in the Trodelvy arm. In addition, Troldelvy demonstrated an effect in controlling symptoms and pain caused by cancer and improvement in patients' quality of life by improving their overall health status. Trodelvy was awarded the highest possible score of 5 points on ESMO-MCBS, the European Society for Medical Oncology's (ESMO) scale used to rate the value of anticancer drugs. A score of 5 indicates that a drug is effective not only in prolonging patient survival but also in improving quality of life, and Trodelvy is the only treatment for metastatic triple-negative breast cancer to receive a score of 5 on ESMO-MCBS.

- Company
- MSD expands domestic clinical trial cooperation
- by Whang, byung-woo May 20, 2025 06:00am
- MSD Korea has broken its record for the most clinical trial approvals in Korea and is now in full swing, developing innovative new drugs for Koreans. With open innovation playing an increasingly important role in new drug research and development (R&D), MSD is expanding its ties with Korea, which plays a pivotal role in its global clinical trials. On the 19th, MSD Korea held R:IM (Notification) DAY and highlighted changes in R&D trends under the theme of “A New Paradigm in the Pharmaceutical Industry: Global Clinical Trends and MSD's Vision.” Korea plays a pivotal role in global MSD cancer research The reason why MSD Korea's performance in the domestic R&D field is attracting attention is that it received the most clinical trial approvals last year. In 2024, MSD Korea received 36 clinical trial approvals from the Ministry of Food and Drug Safety, the most among domestic pharmaceutical companies. During the same period, AstraZeneca Korea (22), AD pharma (19), AbbVie Korea (17), and Boehringer Ingelheim Korea (15) received clinical trial approvals from the MFDS. In particular, MSD Korea has been leading Korea’s medical ecosystem by investing more than KRW 70 billion for 4 consecutive years since 2021, with a cumulative total of KRW 290 billion in research and development costs. This proves the company’s R&D competitiveness in terms of the number of clinical trials, costs, as well as its quality and quantity. Hyunjoo Lee, Executive Director of Clinical Research at MSD Korea Hyunjoo Lee, Executive Director of Clinical Research at MSD Korea, who made a presentation on that day, said, “We are focusing on research and development of new drugs that have been proven to be effective and safe for Koreans in cooperation with domestic research institutes and academic societies.” MSD Korea has also been playing a central role in MSD's global anticancer drug clinical trials. Currently, the company is conducting over 180 clinical trials in collaboration with more than 640 domestic research institutions, with the largest portion (161 studies) focused on oncology. Although Korean institutions account for only 3% (518) of the 14,770 global institutions conducting anticancer drug clinical trials, these institutions are responsible for 73% of MSD's global cancer drug trials, demonstrating the concentrated utilization of Korea's research capabilities. Major hospitals in Korea, such as Seoul National University Hospital, Samsung Medical Center, Asan Medical Center, and Severance Hospital, are among the top institutions leading MSD's global clinical trials, which is considered a testament to Korea's internationally recognized clinical research capabilities. In addition, Korea ranks fourth in the world in terms of the number of patients enrolled in MSD's global anticancer drug clinical trials, following the United States, China, and Japan. Accelerating digitalization... challenge remains on establishing a sustainable cooperation model Another trend in the global pharmaceutical industry's clinical trials is the integration of digital technology. MSD is also actively promoting the introduction of digital technology and artificial intelligence (AI) to keep pace with this trend. A representative example is the increased use of innovative clinical designs such as umbrella, basket, and adaptive protocols in clinical trial design. “MSD is systematically introducing AI and machine learning technologies into the drug discovery and development process to accelerate pipeline diversification,” said Lee. ”MSD has developed AI tools that enable more accurate evaluation of the safety and efficacy of the active substances in its pipeline during the preclinical development stage.” MSD Korea In the long term, MSD Korea aims to become a trusted R&D partner, a bridgehead for domestic pharmaceutical and biotech companies to gain global research experience and enter the market, and foster an ecosystem for innovative new drug development. This includes improving the regulatory environment in Korea, expanding clinical trial infrastructure, and building more active partnerships with domestic research institutions and biotech companies. Currently, MSD Korea is expanding its partnership by conducting joint clinical trials with major domestic biotech companies for the combination therapy of the immuno-oncology drug Keytruda. In the future, the company plans to expand this collaboration to various other fields to build a sustainable ecosystem that will enhance the global competitiveness of the Korean pharmaceutical industry. Lee stated, “To enhance the global competitiveness of Korea's pharmaceutical and biotechnology industry, it is essential for the government, academia, and industry to collaborate closely. We will strive to establish a sustainable collaboration model to position Korea as a global hub for pharmaceutical research.”
- Policy
- Citus generics price raised, Ameliebou Inj huge price cut
- by Lee, Tak-Sun May 20, 2025 06:00am
- Prices of Citus generic drugs will be raised. As the price of original Citus has been adjusted, the prices of generic drugs that were reimbursement-listed in January have been recalculated. Meanwhile, the price of Samsung Bioepis' Lucentis biosimilar 'Ameliebou Inj' has been substantially cut, resulting in a significant difference from the original. According to industry sources on May 19, the prices of some items, including Citus generics, will be adjusted on the 1st of next month. Four Citus (Pranlukast Hydrate) generic products will see price increases following applications to adjust their upper-limit price. The price of Dasan Pharmaceutical's 'Prituss Tab 50 mg,' which met all the criteria, will rise from KRW 344 to KRW 526. In comparison, Daewoong Bio's 'Cituone Tab 50 mg,' Green Cross' 'Neopran 50 mg,' and DongKook Pharmaceutical's 'Pranpid 50 mg,' each meeting only one criterion, will increase from KRW 263 to KRW 447. Prices of Citus generics have been increased: 1. At the the Health Insurance Review and Assessment Service (HIRA)'s Drug Reimbursement Evaluation Committee (DREC) meeting held on April 3, the committee accepted the manufacturers' objection to HIRA's mandatory price reduction of Citus Tab 50 mg. SAMA Pham argued that its Citus development reference product was not the one identified by HIRA and that the ceiling price of Citus had already been adjusted to 53.55%. The committee accepted this argument, and the mandatory cut was effectively canceled. Because the generics listed in January were also assumed never to have had their ceiling price adjusted, their prices were calculated at 53.55% of the highest price for the same formulation. However, when HIRA acknowledged its error in using the original's price-setting criteria and launched a recalculation procedure, the generic prices were adjusted as well. In addition, after price-adjustment applications and negotiations with the National Health Insurance Service, the prices of two Custodiol Solution (Zenith Pharm) products will be raised. The 1,000 mL formulation will rise from KRW 140,492 to KRW 161,981, and the 5 L formulation from KRW 703,347 to KRW 809,905. Ceiling prices of six products will be loswered at the manufacturers' voluntary reduction request. Among these, the dramatic cut for the Lucentis biosimilar 'Ameliebou Inj 10 mg' (Samsung Bioepis), is notable. Ameliebou Inj was listed for reimbursement at KRW 463,773 in January 2023 but was cut to KRW 350,000 that March. It has now been reduced further to KRW 150,000. Since the original Lucentis 10 mg is priced at KRW 579,716, the price gap has widened even more. However, with competitor Chong Kun Dang's LucenBS also set at KRW 150,000, competition between the two drugs is expected to be intense. Ameliebou Inj is currently marketed domestically by Samil Pharmaceutical. Voluntary price cut items (as of June 1): 1. Yungjin Pharmaceutical's Demenduo Tab 10/20 mg (Donepezil Hydrochloride Monohydrate+Memantine Hydrochloride), combination therapy for dementia, will be cut from KRW 3,879 to KRW 3,650. Of the eight reimbursement-listed combination therapies approved in March, three have already cut their prices, indicating fierce price competition.

- Policy
- MFDS to review reference drug application every 2 months
- by Lee, Hye-Kyung May 20, 2025 05:59am
- The change in the procedure for selecting reference drugs for pharmaceutical equivalence tests is being well received by the domestic pharmaceutical industry. The Ministry of Food and Drug Safety recently announced a revision to the 'Guideline for Selecting Reference drugs for Pharmaceutical Equivalence Tests' and is seeking opinions on the change, deciding to proceed with the application and announcement of reference drugs every 2 months instead of every quarter. With the revision of the guideline, reference drug applications will be accepted six times a year (September 16-November 15, November 16-January 15, January 16-March 15, March 16-May 15, May 16-July 15, and July 16-September 15) and reference drugs will be announced in February, April, June, August, October, and December, respectively. The shortened period for selecting reference drugs is expected to help facilitate the smooth conduct of pharmaceutical equivalence tests and the development of generic drugs. In this regard, an official from pharmaceutical company A said, “In the past, the delay in designating a reference drug has often delayed the development schedule. If the designation of a reference drug is regularly conducted two months rather than every quarter, this will well speed up development.” Another official from pharmaceutical company B also said that shortening the schedule for selecting and announcing reference drugs will basically revitalize generic drug development, which is why domestic companies cannot be opposed to it. However, the official added that in practice, shortening the reference drug designation by 2-3 months cannot significantly reduce the drug development period. He emphasized, “We also expect improvements in operational methods, starting with the revision of the reference drug selection and announcement procedures. While designating reference drugs is important, it is also necessary to make reasonable judgments on whether to maintain the reference drug status for drugs that are not imported or produced domestically, despite being designated as reference drugs.” When reviewing applications for the designation (or change) of reference drugs, the government selects products from the same active ingredient type and content, formulation, and route of administration among approved (or reported) products, but those of a higher priority. The priority order for selecting reference drugs is as follows: ▲New drugs as defined in Article 2 of the Pharmaceutical Affairs Act that have received approval as prescription drugs for manufactured (imported) products; ▲Products by the original developer (if there are multiple products, the product with the earliest approval date); ▲Products that are subject to data submission under Article 2, Paragraph 8 of the Regulations on the Approval, Notification, and Review of Drug Products, and are the first products approved in Korea; ▲Products that meet Item 1 or Item 2 and have undergone bioequivalence testing ▲Domestically first-approved products (if the product has been canceled or withdrawn, it refers to products with the same active ingredient type and administration route as the domestically first-approved product, following the order below) etc. A representative from Company B stated, “If there is no reference drug, the reference drug must be purchased from overseas to conduct equivalence tests, but the company whose drug was designated as the reference drug does not disclose all details, such as the quantity of the active pharmaceutical ingredient. While the selection and announcement of reference drugs are important, the system must be improved to ensure proper operation, including measures to revoke reference drug status.”
- Company
- Hanmi-MSD collaborate for R&D
- by Cha, Jihyun May 20, 2025 05:59am
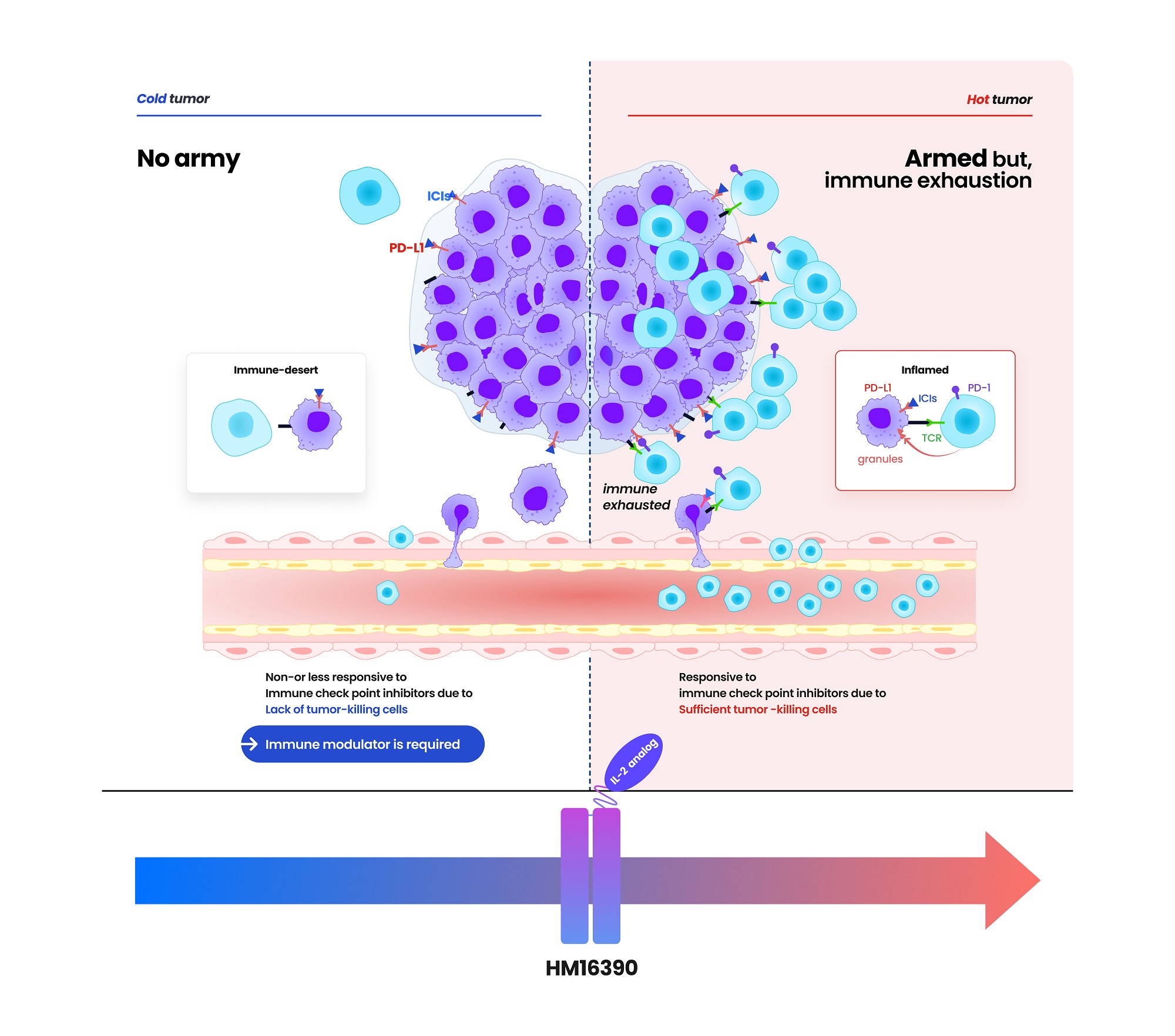
- Hanmi Pharmaceutical entered into a clinical trial collaboration agreement with the U.S.-based Merck (MSD) for developing an immune anticancer drug candidate. The clinical collaboration between Hanmi Pharmaceutical (hereafter, Hanmi) and Merck has expanded to three cases. In addition to clinical trial collaboration, Hanmi continues to collaborate with MSD for efforts such as technology transfers. According to pharmaceutical sources on May 20, Hanmi recently signed a clinical collaboration and distribution agreement with MSD to evaluate the combination therapy containing 'HM16390,' a next-generation anticancer drug that is "LAPS interleukin-2 (IL-2) analog," and MSD's anti-PD-1 anticancer drug 'Keytruda (active ingredient name: pembrolizumab). According to the agreement, Hanmi will be responsible for conducting the Phase 1 trial to assess the safety and efficacy of HM16390+Keytruda combination therapy as a clinical trial sponsor. MSD will supply Keytruda used in clinical trials. HM16390 is a next-generation interleukin-based immuno-oncology candidate that activates immune cells. HM16390 induces T-cell proliferation and activation to enhance immune responses within the tumor microenvironment. By increasing the number of tumor-infiltrating lymphocytes (TIL) that respond to immune checkpoint inhibitors in the tumor microenvironment, it is designed to convert 'cold tumors' (with low immune activity) into 'hot tumors' (infiltrated by immune cells) and to maximize efficacy when used in combination with checkpoint inhibitors. 'Proleukin' is the only recombinant IL-2 therapy approved by the U.S. Food and Drug Administration (FDA). Still, its use is limited due to the risk of adverse events at high doses. Most IL-2 analogs in development focus on enhancing binding affinity to the IL-2 β-receptor to boost anti-tumor effects, which can trigger excessive systemic immune responses and lead to severe side effects such as cytokine release syndrome. (source: Hanmi Pharmaceutical) Hanmi has addressed these limitations by finely adjusting HM16390's binding affinity to the IL-2 α-receptor. According to Hanmi, this approach secures safety while maximizing efficacy. In particular, Hanmi is developing HM16390 as a long-acting therapeutic using its proprietary Labsccovery platform technology, enabling once-per-cycle subcutaneous (SC) administration alongside chemotherapy. HM16390 is currently in a multinational Phase 1 clinical trial. Earlier, Hanmi announced at the Society for Immunotherapy of Cancer (SITC) last November that preclinical studies of HM16390 demonstrated complete remission. Keytruda, MSD's immuno-oncology agent, is the world's top-selling pharmaceutical. It is an immune checkpoint inhibitor that blocks the interaction between PD-1 on T cells and PD-L1 on cancer cells, enabling immune cells to attack tumor cells. First approved by the U.S. FDA in September 2014 for malignant melanoma, Keytruda has continuously added new indications. It has over 40 indications to date, including breast, gastric, and lung cancers, making it the checkpoint inhibitor with the broadest range of cancer uses. However, the efficacy of Keytruda is limited to patients whose tumors express high levels of PD-L1. It is known that those with low PD-L1 expression derive minimal benefit. To address this, MSD is actively developing combination therapies to expand the responsive patient population and enhance efficacy. Hanmi expects that combining HM16390 with Keytruda will further improve treatment outcomes. Hanmi and MSD have expanded collaboration deals to three in total. Hanmi also conducts Keytruda combination therapy clinical trials with MSD's PD-L1/4-1BB bispecific antibody candidate 'BH3120' in combination with its oral CCR4 antagonist, 'Tivumecirnon.' BH3120, which is being co-developed by Hanmi and Beijing Hanmi Pharm, is a bispecific antibody immuno-oncology candidate in which a single antibody simultaneously binds to two different targets. It features Hanmi's proprietary Pentambody platform technology, combining an immuno-oncology mechanism that activates immune cells with the target-oncology characteristic of selectively attacking only cancer cells. In particular, Hanmi explains that BH3120 is designed to respond differently to each target, PD-L1 and 4-1BB, thereby enhancing therapeutic efficacy while reducing side effects. Hanmi plans to present the interim results of the Phase 1 clinical trial evaluating the BH3120 and Keytruda combination therapy in the second half of this year. Previously, Hanmi Pharmaceutical reported that in tumor-bearing mouse models refractory to immune-oncology agents, the combination of BH3120 and Keytruda demonstrated tumor growth inhibition that was at least comparable to that observed with a competing pipeline agent (GEN1046). R&D new pipeline: Preclinical trials, Phase 1 Trials, Phase 2 Trials, Phase 3 Trials, Approved (source: Hanmi Pharmaceutical) Tivumecirnon is an oral immuno-oncology candidate Hanmi acquired from RAPT Therapeutics in 2019. It blocks the CCR4 receptor protein, thereby inhibiting the migration of regulatory T cells that suppress immune responses into tumors. In January, at the ASCO Gastrointestinal Cancers Symposium held in San Francisco, Hanmi presented a poster on the Phase 2, Part 1 results for Tivumecirnon. In collaboration with RAPT and MSD, this trial treated ten Epstein-Barr virus (EBV)-positive gastric cancer patients and achieved an objective response rate (ORR) of 60%. ORR is a key efficacy metric representing the proportion of patients whose tumors have either disappeared entirely or shrunk by a defined amount following cancer treatment. Among these responses, there was one complete response and five partial responses. The median time to response (mTTR) was 2.7 months. The median duration of response (mDOR) was 17.3 months. In Cohort 2, the median progression-free survival (PFS) was 10.4 months. Hanmi explained that the treatment-related adverse events observed among the 20 patients enrolled in the trial were mostly manageable. In addition to its clinical collaboration with MSD, which is focused on Keytruda combination therapy, Hanmi is also maintaining partnerships via technology licensing agreements. For instance, it includes 'efpeglenatide,' which Hanmi licensed to MSD in 2020 in a deal valued at USD 860 million. Efpeglenatide is a dual-action agent that activates the glucagon-like peptide-1 (GLP-1) receptor, which enhances insulin secretion and suppresses appetite, and the glucagon receptor, which increases energy metabolism. Hanmi previously licensed efpeglenatid to Janssen in 2015 for obesity and diabetes indications, regained the rights in 2019, repurposed it for metabolic-associated steatohepatitis (MASH), and successfully licensed it again to MSD. MSD is currently conducting a Phase 2 clinical trial of efpeglenatide. The trial compares efpeglenatide with the comparator treatment, semaglutide from Novo Nordisk, and a placebo. According to a 2023 presentation at the European Association for the Study of the Liver (EASL) in Vienna, data from the Phase 2a analysis showed that at week 24 of treatment, efpeglenatide reduced liver stiffness by 72.7% compared to baseline. This result markedly outperformed semaglutide, which achieved a 42.3% reduction over the same period. Resolution of steatosis without fibrosis worsening and improvement of fibrosis without steatosis worsening are key evaluation endpoints defined by the FDA for NASH therapies. MSD aims to complete the Phase 2 trial of efpeglenatide by December of this year.
- Company
- Hanmi partners with MSD for next-gen IL-2 analog development
- by Cha, Jihyun May 20, 2025 05:58am
- Hanmi Pharmaceutical (CEO: Jae-Hyun Park) announced on the 19th that it has signed a clinical trial collaboration and supply agreement with U.S. Merck (MSD) to evaluate the combination therapy of its LAPS IL-2 analog 'HM16390' and MSD's anti-PD-1 immunotherapy 'Keytruda' (pembrolizumab). Hanmi Pharmaceutical will sponsor and oversee the Phase I clinical trial to evaluate the safety and efficacy of the combination therapy of HM16390 and Keytruda. MSD will supply Keytruda for the clinical trial. HM16390 is a next-generation IL-2 variant designed with a differentiated strategy that regulates the differentiation and proliferation of immune cells. HM16390 is designed to maximize antitumor effects by increasing the number of tumor-infiltrating lymphocytes that respond to immune checkpoint inhibitors in the tumor microenvironment through a mechanism that induces T cell proliferation and activation, thereby converting cold tumors with low immunogenicity into hot tumors with high immunogenicity. Currently approved recombinant IL-2 therapy 'Proleukin' is recommended for limited use due to side effect issues. Additionally, most IL-2 analogues in development focus on regulating the binding affinity of the IL-2 beta receptor, but this approach has shown limitations in terms of safety, according to Hanmi Pharmaceutical. Reducing the binding affinity of the IL-2 beta receptor decreases side effects such as vascular leak syndrome, but this also reduces anticancer effects. Conversely, increasing the binding affinity of the IL-2 beta receptor and eliminating binding with the IL-2 alpha receptor enhances anticancer effects, but this increases the risk of severe side effects such as cytokine release syndrome. To overcome these limitations, Hanmi Pharmaceutical has introduced a differentiated development strategy for HM16390. Unlike existing IL-2 candidates, HM16390 precisely regulates the binding affinity of the IL-2 alpha receptor, thereby ensuring safety while maximizing the efficacy of the drug. The company expects that this approach will maintain anticancer effects while minimizing serious side effects. (Data: Hanmi Pharmaceutical) HM16390 is an immunotherapy drug that maximizes the efficacy, safety, and durability by applying Hanmi Pharmaceutical's proprietary sustained-release platform technology, LAPSCOVERY. It is currently being developed as a sustained-release therapy that can be administered once per treatment cycle via subcutaneous injection (SC). Hanmi Pharmaceutical is developing HM16390 for use as a monotherapy and in combination with other immunotherapy agents for various solid tumors, and is currently conducting a global Phase I clinical trial. Dr. Jong Chul Park, Professor at the Massachusetts General Hospital (MGH) Head and Neck Cancer Center, Harvard Medical School, and the principal investigator for the Phase I clinical trial of HM16390 in Korea and the United States, said, “Through collaboration with MSD, we anticipate that the combination therapy of HM16390 and Keytruda will improve treatment outcomes for patients with advanced or metastatic solid tumors, and expect significant results in the future.” Young Su Noh, Director of Hanmi's ONCO Clinical Team, said, “Hanmi Pharmaceutical possesses a differentiated pipeline in the field of oncology, particularly in immunotherapy. We plan to sequentially showcase our research achievements through various academic conferences this year.”
- Company
- 'Oxlumo' for primary hyperoxaluria expected to be available
- by Eo, Yun-Ho May 19, 2025 05:56am
- The primary hyperoxaluria treatment, 'Oxlumo,' is expected to be commercialized in South Korea. According to sources, the Ministry of Food and Drug Safety (MFDS) reviewing Oxlumo (lumasiran) for approval. The MFDS granted 'Global Innovative products on Fast Track (GIFT)' designation to Oxlumo last year and orphan drug status in October of the same year. Oxlumo is an RNAi therapy for primary hyperoxaluria type 1 (PH1), a rare kidney disease, that was approved by the U.S. Food and Drug Administration (FDA), and the European Medicines Agency (EMA) in 2020. RNAi is one of the gene therapies considered a next-generation new drug technology, and it provides an advantage for specifically targeting human genes that cause diseases. PH1 is a rare disease in which the liver produces excessive oxalate. It causes the accumulation of oxalate crystals or calcium oxalate in the liver and urinary system. When the disease continues, the kidneys are damaged, requiring kidney dialysis. A treatment option for PH1 became available with the approval of Oxlumo in 2020. Oxlumo is an RNAi therapy targeting hydroxyacid oxidase 1 (HAO1), coding the oxalate-producing glycolate oxidase (GO) enzyme. It works by suppressing HAO1 and reducing GO production, ultimately reducing oxalate levels. Meanwhile, the efficacy of Oxlumo was found in a Phase 3 study involving 39 PH1 patients aged six years or older. Patients treated with Oxlumo had 65.4% lower oxalate levels in urine compared to the placebo group. Furthermore, 84% of patients treated with Oxlumo had oxalate levels close to normal. 52% of the group had recovered to the normal range.
- Opinion
- [Reporter's View] Strengthening GMP for sterile drugs
- by Kim, Jin-Gu May 19, 2025 05:56am
- The government has reaffirmed its existing position on strengthening GMP standards for sterile finished drug products in December. The Ministry of Food and Drug Safety recently met with sterile drug factory managers and stated that there will be no postponement of the enforcement of the “Regulations on Drug Manufacturing and Quality Control” in December. Under these regulations, facilities producing sterile drug products must implement the following measures: ▲Establish and implement a systematic contamination control strategy for the manufacture of sterile drug products; ▲Develop individual good manufacturing practices (GMP) for advanced biopharmaceuticals; ▲Clarify the details of the specific dosage forms, evaluation procedures, and methods for determining compliance with the good manufacturing practices (GMP). The MFDS has been preparing for the implementation of these measures since joining the Pharmaceutical Inspection Co-operation Scheme (PIC/S) in 2014. The MFDS believed that it was necessary to strengthen GMP standards for sterile products in line with international standards. To this end, the MFDS has provided the pharmaceutical industry with sufficient time. When revising relevant regulations in 2023, the MFDS required manufacturers of sterile finished drug products to replace outdated equipment within 2 years after the revision of the notice, and manufacturers of sterile APIs to do so within three years. Additional grace periods were granted for certain provisions. However, as the deadline for implementing the new regulations approaches, an increasing number of pharmaceutical companies are refusing to replace their outdated equipment. Instead of halting production of sterile drug products, they plan to shift production to contract manufacturing facilities. This is because replacing outdated equipment could cost tens of billions to hundreds of billions of won, depending on the facility. At first glance, this seems as if aseptic drug manufacturers are considering production halts to save equipment replacement costs. Additionally, given the sufficient timeframe provided, it seems that they have not been reacting for the past two years and only decided to halt production as the deadline for stricter regulations approaches. However, the story of sterile drug manufacturing plants is different. Their common claim is that the productivity of sterile drug manufacturing itself has become too low, regardless of the need to replace outdated equipment. In a situation where profits are almost nonexistent due to excessively low drug prices, they argue that they have no choice but to halt production, as additional costs of up to hundreds of billions of won would be required for replacements. For this reason, the pharmaceutical industry requested support for facility investment costs and improvements to the drug price structure during several recent meetings with the MFDS. However, the MFDS has drawn a line, saying that it will not provide investment support as it has already provided a sufficient grace period. Discussions on pricing sterile drugs have also stalled. If this situation continues, there are concerns that the supply shortage of sterile drug products will worsen by the end of the year. Already this year, 22 cases of injection supply discontinuations or shortages have been reported. It is known that about 10 sterile drug factories are seriously considering discontinuing production. If they discontinue their product supply by the end of the year, there are concerns that it will lead to a large-scale supply shortage. I agree with the policy direction that GMP should be strengthened to meet international standards. However, before doing so, we must first address the fundamental problem of low productivity. The MFDS must not dismiss the demands of sterile drug product manufacturers as “lack of preparation.” Unless the fundamental problems of excessively low drug prices and the resulting low productivity are resolved, it will be difficult to encourage sterile drug product manufacturers to participate, no matter how long the grace period is.