- LOGIN
- MemberShip
- 2026-06-22 16:59:21
- Company
- 'Voranigo' for malignant brain tumor expected to land in KOR
- by Eo, Yun-Ho Nov 11, 2024 05:49am
- Product photo of Voranigo. 'Voranigo,' a new therapy for malignant brain tumors in over twenty years, is anticipated to be brought to South Korea. Sources said that Servier has recently filed an approval application for Voranigo (vorasidenib). Earlier in September, the Ministry of Food and Drug Safety (MFDS) assigned Voranigo an Orphan Drug Designation (ODD). Voranigo's indication is to treat diffuse gliomas with an isocitrate dehydrogenase (IDH) mutation. Voranigo targets patients with low-grade gliomas who are vulnerable to IDH1 or IDH2 mutations. This drug is a bispecific inhibitor of IDH1/2, developed by multinational pharmaceutical company Servier. It targets glioma (astrocytoma or oligodendroglioma), a type of brain tumor that is difficult to treat. Voranigo received the final approval from the U.S. Food and Drug Administration (FDA) in August and is in the process of getting approved in countries worldwide, including Europe. The efficacy of Voranigo was demonstrated through the Phase 3 INDIGO study. The study results were presented at last year's American Society of Clinical Oncology (ASCO) Annual Meeting. The study results showed that in patients with glioma who have IDH mutation, the drug significantly reduced the tumor progression or death risk by 61% compared to the placebo. Voranigo also reduced the risk of requiring further treatments by 74%. The study participants had no history of undergoing treatments other than surgery. Voranigo also showed favorable drug tolerance. Its safety profile was similar to the Phase 1 clinical trial case results. Meanwhile, glioma is the most common type of primary malignant brain tumor in adults, and almost all adult patients (grade 2 diffuse gliomas) are likely to have IDH1 or IDH2 mutation. These patients are currently treated with off-label (medicines approved for other uses) instead of officially approved medicines. In addition to Voranigo monotherapy, Servier is currently conducting clinical trials for concurrent use of the drug with MSD's immune checkpoint inhibitor, 'Keytruda (ingredient: pembrolizumab),' in patients with relapsed and advanced gliomas who have IDH1 mutation. The company already has 'Tibsovo (IDH1 inhibitor)' and an IDH2 inhibitor in its IDH inhibitor pipeline.

- Company
- K-Bios showcases new ADC drugs on the international stage
- by Son, Hyung Min Nov 11, 2024 05:49am
- The domestic pharma and biotech industry is speeding up the development of new antibody-drug conjugates (ADCs), unveiling their clinical results. At the 'World ADC 2024' that was held in San Diego, U.S. for 4 days from the 4th of this month, domestic pharmaceutical bio companies including Celltrion, LigaChem Biosciences, and ABL Bio revealed ADC research results. With the results, the companies are aiming to secure various solid cancer indications such as non-small cell lung cancer, bladder cancer, and breast cancer. Celltrion seeks to develop a new ADC drug with the Pinotbio platform According to industry sources on the 9th, Celltrion unveiled the preclinical results of its ADC candidate 'CT-P70' for non-small cell lung cancer and 'CT-P71' for bladder cancer and other solid cancer indications for the first time at the conference. The new pipeline unveiled at World ADC utilizes PBX-7016, an ADC platform developed by the ADC company Pinotbio through open innovation. The platform technology is characterized by its enhanced hydrophilicity to increase stability in the blood through the jointly developed payload 'camptothecin derivative,’ and has excellent anti-cancer effects on tumors. ADCs are anticancer drugs made by connecting antibodies that bind to specific target antigens on the surface of tumor cells to cytotoxic payloads with a linker, enabling the drug to selectively act only on cancer cells. This offers the advantage of enhancing therapeutic efficacy while minimizing side effects. With ADC drugs such as Daiichi Sankyo and AstraZeneca's ‘Enhertu,’ Gilead's 'Troldelvy', and Takeda's 'Adcetris' successfully settling in the market, the industry is eyeing the emergence of follow-up drugs. Currently, technology transfer and clinical trials for the next ADC are actively underway in global and domestic pharmaceutical companies. CT-P70 is an ADC drug targeting solid cancers such as non-small cell lung cancer. It targets 'c-MET', which causes tumor growth when activated in cancer cells. C-MET is a protein expressed by the epithelial-to-mesenchymal transition (MET) gene. It is one of the proteins that transmit signals to cells and is considered a typical cancer-inducing gene and is associated with the development of various solid cancers, including colorectal, gastric, and liver cancers, as well as lung cancer. It is estimated that 6% of patients with NSCLC have c-MET mutations. In preclinical studies, CT-P70 has shown tumor inhibition effect in c-MET-expressing lung and gastric cancers in vitro and in vivo, with confirmed safety in toxicity studies. There are currently no ADCs targeting c-MET available on the market, but AbbVie presented promising Phase II trial results last year. Based on the Phase II trial results, AbbVie has applied for accelerated approval with the U.S. Food and Drug Administration (FDA). CT-P71, which Celltrion also presented the same day, is an ADC drug in development for the treatment of solid tumors, including bladder cancer, that targets nectin-4, a cell surface protein that is overexpressed in solid tumors. It has shown efficacy in tumor inhibition in bladder, breast, and lung cancers in nonclinical studies and an excellent safety profile in toxicity studies. Currently, the only ADC drug that targes nectin-4 is Astellas' Padcev. The drug is currently approved for urothelial carcinoma and is being studied in several solid tumors. Celltrion plans to develop CT-P70 and CT-P71 as best-in-class drug candidates. ABL Bio-LigaChem Biosciences announces ADC clinical trial results ABL Bio and Ligachem Biosciences, which have a diverse pipeline of ADC drug candidates, also attended the event. LigaChem Biosciences unveiled the features and safety results of its ADC platform 'ConjuAll'. ADCs consist of a linker, payload (drug), and antibody, and the ConjuAll linker is said to be able to overcome the release of cytotoxic drugs into the blood and attack normal cells. Currently, LigaChem Biosciences has three drug candidates in clinical trials - LCB14, LCB84, and LCB71. LCB14 is an ADC that targets HER2 and is currently in Phase II/III clinical trials in China and Phase I trials in Australia. LigaChem Biosciences has licensed out LCB14 to China-based Fosun Pharma UK-based Iksuda Therapeutics. The two companies are conducting clinical trials in China and Australia, respectively. LCB84, a Trop2-targeting ADC, was licensed out to Janssen. LCB84 is an ADC drug candidate with the potential to target a variety of solid tumors, including triple-negative breast cancer and non-small cell lung cancer. In preclinical studies, LCB84 has shown efficacy in solid tumors refractory to topoisomerase payload-based TROP2 ADC drugs. LCB71 is currently in Phase I clinical development. LCB71 is an ADC candidate that targets hematologic malignancies. Currently, LigaChem Biosciences’ partner in China, Cstone, has selected LCB71 as its core clinical program and plans to initiate a Phase Ib clinical within the year. ABL Bio presented non-clinical trial data from its bispecific ADC pipeline. The two drug candidates it presented were found to be well-tolerated and safe in preclinical studies. ABL Bio is developing ABL201, an ADC candidate targeting hematologic cancers, and ABL201 and ABL202 targeting solid tumors. ABL has successfully licensed out ABL201 to a biotech venture TSD Life Sciences. Also, ABL202 is being developed in collaboration with LigaChem Biosciences’ linker technology. The company aims to enter clinical trials for three of its bispecific ADCs, including the one in preclinical trials.
- Company
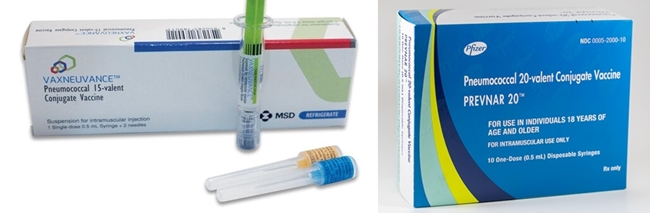
- 20-valent pneumococcal conjugate vaccine to enter market
- by Whang, byung-woo Nov 08, 2024 05:47am
- The pneumococcal vaccine market, which has shifted with the introduction of a 15-valent vaccine this year, is expected to again face fierce competition with the approval of a new 20-valent vaccine. Pfizer, which is about to launch a new product, plans to replace the share of its existing product, Prevenar 13 with its new product, Prevenar 20. In the process, MSD, which has been gaining market share since the launch of Vaxneuvance, will also be facing increasing challenges to secure its sales. (From the left) Product photos of Vaxneuvance and Prevenar 20 According to industry sources on the 7th, the Ministry of Food and Drug Safety recently approved Pfizer's ‘Prevenar 20 Prefilled Syringe' (Prevenar 20). Prevenar 20 is Pfizer's first new pneumococcal vaccine in 14 years, which adds 7 serotypes (serotypes 8, 10A, 11A, 12F, 15B, 22F, and 33F) to the existing Prevenar 13 vaccine. It can be used to vaccinate infants, children, and adolescents aged 6 weeks to 18 years and adults aged 18 years and older. Prevenar 20 is expected to be released in the first half of next year, given that Vaxneuvance was approved a year ago on Oct. 31, 2023, and then released to market in April the following year. A Pfizer spokesperson said, “We are currently working with multiple stakeholders to expedite all processes to accelerate the launch of Prevenar 20. We will share more details as soon as we have a specific launch timeline.” Prevenar 13 was sold in partnership with Chong Kun Dang for adults and Korea Vaccine for pediatric use. Industry insiders believe that these two companies will have priority in discussing the distribution of Prevenar 20. If there is one variable in terms of timing, its entry into the National Immunization Program (NIP) would likely be the variable. Currently, pediatric pneumococcal vaccines are included in the NIP, and both Prevenar 13 and Vaxneuvance are available through the program. Last year, Vaxneuvance passed the Korea Disease Control and Prevention Agency’s Korea Expert Committee on Immunization Practices within a month of its approval, expanding its market influence by applying the pediatric NIP upon its launch. In fact, Jae-Yong Cho, executive director of vaccine business at MSD Korea, said at a recent media seminar that “Both the initial vaccination cases using Vaxneuvance for infants and young children under the NIP, as well as cross-dosing cases where subjects are switching from the existing 13-valent vaccine to Vaxneuvance, have been increasing.” Pfizer is also reportedly in discussions with the KDCA about applying pediatric NIP to Prevenar 20 to gain competitiveness. “Pediatric pneumococcal vaccination covered by the NIP, so Pfizer has no choice but to enter the NIP to make Prevenar 20 competitive,” said a vaccine industry insider. ”We understand that they are currently in discussions with the KDCA, but whether the drug will be included in the NIP will depend on how Pfizer sets the price.” The industry's view is that if the discussions between the KCDA and Pfizer go smoothly, it could lead to a quick launch, but if there is a big disagreement on the price, the pharmacoeconomic evaluation process will be inevitable, changing Pfizer's prompt introduction strategy. Prevenar 13 and Vaxneuvance compete in clinics in Korea “We are making our best efforts to promptly introduce Prevenar 20 into the National Immunization Program. We are working closely with relevant departments to prepare the necessary procedures,” said a Pfizer representative. Apart from the pediatric NIP entry, pricing is also expected to be a key issue in the adult market, as a non-reimbursed launch is certain in the area. In the current market, Vaxneuvance is only slightly more than Prevenar 13. The successor to Prevenar 13, Prevenar 20, is expected to be a little more expensive. Upon its launch, the company had been running TV commercials to build brand awareness for Vaxneuvance. The company has also been emphasizing immunogenicity, which is as important to the vaccine's effectiveness as the scope of protection.
- Company
- Optivo's clinical results for Koreans with gastric cancer
- by Son, Hyung Min Nov 08, 2024 05:47am
- Product photo of the immunotherapy Optivo. Optivo, an immunotherapy used to treat certain types of cancer, showed a positive result in gastric cancer clinical trials that involved Koreans. The study demonstrated a 47% reduction in the disease progression and death risk compared to conventional therapies. According to industry sources on November 6th, Bristol Myers Squibb (BMS) and Ono Pharmaceutical have recently presented the subgroup analysis results from Phase 3 'ATTRACTION-4,' evaluating Optivo as a treatment for East Asian patients. The clinical trial involved 724 patients with HER2-negative gastric cancer, including 291 Korean patients. The study evaluated Optivo in combination with platinum-based chemotherapy as a first-line treatment for patients compared to a combination therapy of placebo plus platinum-based chemotherapy. Based on the clinical result, Optivo combination therapy's benefit of improving progression-free survival (PFS) was prominent in the Korean patient group compared to those from other countries. In detail, the Korean subgroup's median PFS was 14.8 months for Optivo combination therapy and 8.3 months for placebo, showing 47% lower disease progression and death risk with Optivo combination therapy. The objective response rate (ORR) was 54.7% for the Optivo combination therapy group, which was higher than the placebo group's. The median duration of response (DOR) was 16.0 months for the Optivo combination therapy group, which was longer than the 9.9 months for the placebo group. In the total patient group, about 80% of the patients who showed complete response (CR) in the Optivo combination therapy group survived over three years. New safety-related adverse reactions associated with Optivo combination therapy had not been reported. In a retrospective clinical analysis conducted in Asan Medical Center involving patients with advanced gastric cancer who also have advanced deficient mismatch repair (dMMR), patients treated with Optivo combination therapy had a 12-month PFS of 69.4%, which was longer than 20.6% for the placebo group. "The subgroup analysis from the ATTRACTION-4 study, which enrolled many Koreans, showed that Optivo combination therapy yielded positive data during about three years of follow-up. This study results support the treatment effects of Optivo combination therapy, currently listed as a reimbursable first-line treatment for HER2-negative advanced or metastatic gastric cancer," Hyung-Don Kim, a Professor of the Department of Oncology at Asan Medical Center in Seoul, said. Optivo is the only reimbursable drug among immunotherapies…has been established as a standard therapy In South Korea, Optivo was approved in June 2021 as a first-line treatment in combination with fluoropyrimidine- and platinum-containing chemotherapy for patients with advanced or metastatic gastric adenocarcinoma, gastroesophageal junction (GEJ) adenocarcinoma, or esophageal adenocarcinoma. It was listed for reimbursement last year. The average OS was below 1 year for HER2-negative patients, about 80% of advanced·metastatic cancer, who received conventional chemotherapy as a first-line treatment. Compared to immunotherapy for cancer, several targeted therapies used in clinical trials as a first-line treatment had not shown significant clinical benefits. The treatment practices for HER2-negative advanced gastric cancer started to change following the introduction of a treatment strategy of using an immunotherapy agent in combination with conventional chemotherapy. Optivo is now established as a standard therapy after becoming the first immunotherapy to win approval for the indication to treat advanced·metastatic gastric cancer as a first-line treatment in combination with chemotherapy. The basis for Optivo combination therapy used as a first-line treatment of advanced gastric cancer was the Phase 3 CheckMate-649 study, which showed relatively high ORR and longer DRR compared to single chemotherapy in all patient groups, including patients with below PD-L1 CPS 5. Such treatment benefits were repeated in the fourth-year follow-up results presented at the ASCO Gastrointestinal (GI) Cancers Symposium (ASCO GI), held in January. The clinical results showed that the Optivo combination therapy group's below CPS 5 patient group had an ORR of 55%. Optivo combination therapy provided an OS benefit for the patient group accompanying microsatellite instability-high (MSI-H) regardless of the PD-L1 expression rate. "Gastric cancer is a type of cancer with the highest prevalence in South Korea, but Korean patients faced difficulties because of limited treatment option availability," Professor Kim said. "As Optivo combination therapy has demonstrated OS extension in HER2-negative gastric cancer as the first first-line combination therapy, it is meaningful because it provides a treatment option with long-term survival goal." "With reimbursement listing, many patients could benefit from Optivo combination therapy over a year. It might have brought positive changes to the survival rate of advanced or metastatic gastric cancer," Professor Kim added.
- Company
- Geo-Young and UBC Korea sign MOU to distribute Bimzelx
- by Son, Hyung Min Nov 08, 2024 05:47am
- (from the left) Sujin Hwang, General Manager of UBC Pharm, Sun-Hae Cho, Chairwoman of Geo-Young. Geo-Young announced today that it has signed a Memorandum of Understanding with UBC Korea to distribute Bimzelx. Under the agreement, Geo-Young will carry out the entire distribution process from storage to supply of Bimzelx. Bimzelx is a treatment for plaque psoriasis that simultaneously and directly targets and inhibits interleukin 17A and 17F (IL-17A·IL-17F), protein immunomodulators that drive inflammation in psoriatic disease. On August 29, Bimzelx the Ministry of Food and Drug Safety approved Bimzelx as a treatment for adult patients with moderate-to-severe psoriasis who require phototherapy or systemic therapy. Through this distribution agreement, Geo-Young plans to commit itself to ensuring a stable supply of Bimzelx in Korea. In multiple clinical trials, including in South Korea, Bimzelx demonstrated higher levels of Psoriasis Area and Severity Index (PASI) 100 achievement rates than existing biologics. PASI, which stands for Psoriasis Area and Severity Index, is a key efficacy endpoint for psoriasis treatments, and Bimzelx’s PASI 100 achievement rate was maintained at a high level for three years in an open-label extension study. “As the leading drug distributor in Korea, we will do our best to provide prompt and accurate supply of Bimzelx and contribute to the treatment of psoriasis patients,” said Sun-Hae Cho, Chairwoman of Geo-Young. ”We will continue to take initiative in improving public health through the active distribution of excellent medicines.” “We are very pleased to take the first step towards the launch of Bimzelx with Geo-young, following the approval of Bimzelx by the MFDS in August,” said Sujin Hwang, General Manager of UBC Pharm. ”We hope that the stable supply of Bimzelx will help many psoriasis patients in Korea experience the therapeutic benefits of Bimzelx.”
- Opinion
- [Reporter's View] Regarding the obesity drug craze
- by Son, Hyung Min Nov 08, 2024 05:46am
- There is one research and development item that is gaining explosive popularity among both global and domestic pharmaceutical companies –obesity treatment. The use of obesity drugs has also increased dramatically with the significant rise in the number of obese people around the world. Global sales of obesity drugs reached USD 6.68 billion (about KRW 9 trillion) last year, up 145.6% from USD 2.72 billion in 2022. In particular, the success of glucagon-like peptide (GLP-1) receptor agonist obesity drugs such as Saxenda, Wegovy, and Zepbound has attracted the attention of domestic and foreign pharmaceutical companies to GLP-1 drug candidates. Saxenda posted sales of KRW 1.225 trillion last year, up 9.8% YoY. Wegovy’s sales surpassed KRW 300 billion last year despite facing supply difficulties. GLP-1 was originally developed as a diabetes treatment. However, GLP-1 drugs took a major turn when Novo Nordisk's liraglutide and semaglutide showed effect in weight loss. During clinical trials of its antidiabetic drug liraglutide (trade name Victoza), Novo Nordisk noticed a weight loss effect among its enrollees. Based on the findings, the company changed the dose of liraglutide and developed Saxenda, a GLP-1 drug for obesity. The company used the same mechanism to develop Wegovy with semaglutide (Ozempic). Both Saxenda and Wegovy are now in the global market, and their non-reimbursement prices have surged with their increasing popularity. The problem is that the promise of effective weight loss through simple injections has led to increased use for cosmetic and dieting purposes. There has been a significant increase in prescriptions for weight loss in people who are not overweight and obese or chronically ill. The popularity of dietary supplements with unproven efficacy and side effects is also on the rise. However, manufacturers only highlight the weight-loss benefits of GLP-1 drugs and neglect to warn consumers about their side effects. GLP-1 receptor agonists are associated with various side effects. Side effects of GLP-1 class drugs include muscle loss, acute kidney disease, nausea, vomiting, and diarrhea. In the United States, a recent case of acute pancreatitis was reported as a side effect of an anti-obesity drug, resulting in death. Unlike diabetes, obesity is not classified as a disease and is therefore treated without reimbursement benefits. Although it is classified as a risk factor for various metabolic diseases, such as dyslipidemia and hypertension, it is not regarded as a condition that requires medication. The first treatment recommended for obese patients who visit an endocrinologist is lifestyle modification. If the weight remains uncontrolled after lifestyle modification, doctors will consider using a medication, but they are cautious about doing so because of the side effects that come with it, including rebound weight gain. While the overprescription of obesity drugs for cosmetic purposes is a serious problem, the lack of information on its side effects seems to be a major contributor to the misuse and abuse of obesity drugs. The role of the pharmaceutical industry, which has developed and is developing obesity drugs, is also important. While it is necessary to publicize how much weight loss is achieved with the use of each drug, it is also time for the public to be fully informed about their side effects. “Miracle drugs” that can dramatically reduce weight are not free from side effects.

- Company
- Novo Nordisk tops KRW 14T in sales, thanks to Wegovy
- by Whang, byung-woo Nov 08, 2024 05:46am
- Novo Nordisk has recorded a strong sales performance in Q3, driven by the steady growth of its major products, including Wegovy and Ozempic. According to Novo Nordisk's business reports on November 6th (local time), they recorded US$10.267 billion (KRW 14.3378 trillion) in Q3 sales, up 21% from the previous year. Its operating profit amounted to US$4.877 billion (KRW 6.8112 trillion), up 26% from 2023, and it recorded US$3.9325 billion (KRW 5.4921 trillion) in net revenues, up 21% from 2023. By region, the sales rose by 22% to US$3.85297 billion (KRW 5.4861) in the North American market and by 21% to US$2.59707 billion (KRW 3.6275 trillion) in the global market, excluding the North American market. It recorded cumulative sales of US$29.483 billion (KRW 41.1818 trillion) up to Q3 2024. Sales growth of 23-27% and an operating profit increase of 21-27% for the fiscal year 2024 are forecasted. The company analyzed increased sales across the entire portfolio contributed positively to the sales growth in Q3. By products, entire GLP-1 agents recorded sales of US$446.59 million (KRW 623.5 billion), up 14%. Among these, GLP-1 drugs for obesity showed a growth rate of 55%, and GLP-1 drugs diabetes showed a growth rate of 15%. During the same period, rare disease treatments and insulin agents recorded sales growth of 17% (KRW 577.7 billion) and 10% (KRW 1.5805 trillion), respectively. Also, its key product Wegovy (semaglutide), an obesity drug, generated US$2.4997 billion (KRW 3.49 trillion), which rose 79% from US$873 million (KRW 1.2194 trillion) from year-over-year (YoY). The diabetes drug Ozempic generated sales of US$7.82581 billion (KRW 10.9287 trillion) in Q3, up 32% YoY. In contrast, Saxenda (liraglutide), an obesity drug, recorded sales of US$135.45 million (KRW 189.1 billion), down 43% YoY, due to Wegovy's success. Victoza, a diabetes drug, also recorded a negative growth of -41%. "We have achieved significant sales growth due to the rising demand for GLP-1 treatment targeting diabetes and obesity," Lars Fruergaard Jørgensen, CEO of Novo Nordisk, said. Meanwhile, Novo Nordisk announced they have confirmed Wegovy's effect in patients with metabolic dysfunction-associated steatohepatitis (MASH) through the latest ESSENCE study. Based on these clinical trial results, the company will file for approval from the United States and EU in the first half of 2025.
- Company
- Oral psoriasis drug 'Sotyktu' lands at Big 5 hospitals
- by Son, Hyung Min Nov 08, 2024 05:46am
- Product photo of Oral psoriasis drug 'Sotyktu' is now available for prescription at tertiary general hospitals. According to industry sources, BMS Korea’s TYK2 inhibitor, Sotyktu (deucravacitinib), has passed the drug committees of the 'Big 5' tertiary hospitals, including Samsung Medical Center, Seoul University Hospital, Seoul St. Mary's Hospital, Seoul Asan Medical Center, and Sinchon Severance Hospital. Prescription code is also available in major hospitals nationwide, including Gangnam Severance Hospital, Konyang University Hospital, Soonchunghyang University Bucheon Hospital, Seoul National University Bundang Hospital, Soonchunghyang University Hospital Seoul, Ewha Womans University Medical Center, Chosun University Hospital, and Soonchunghyang University Cheonan Hospital. Since being added to the reimbursement list in April, this drug is increasingly available in hospitals. Sotyktu is the first TYK2 inhibitor approved for adults with moderate-to-severe plaque psoriasis. It was approved in August 2023 in South Korea to treat adult patients with moderate-to-severe plaque psoriasis who are candidates for phototherapy or systemic therapy. The insurance coverage was made available eight months after receiving approval. It is covered by insurance reimbursement for the treatment of patients over 18 with chronic severe plaque psoriasis who have symptoms lasting over six months. Coverage requirements include ▲Plaque psoriasis with over 10% of the total skin areas ▲Patients who have Psoriasis Area and Severity Index (PASI) score of over 10 ▲Patients who have undergone methotrexate or cyclosporine for over 3 months but cannot continue the treatment due to no response or side effects ▲Patients who have undergone light therapy or UPV photo therapy for over 3 months but cannot continue the treatment due to no response or side effects. Clinical efficacy of Sotyktu was demonstrated in Phase 3 POETYK PSO-1 and POETYK PSO-2 clinical studies, which compared the drug with placebo or Otezla in 1,684 adult patients 18 years or above with plaque psoriasis. POETYK PSO-1 studies have shown that the Sotyktu treatment group had a PASI 75 response rate of 58.4% at week 16, which was significantly higher than the apremilast group's 35.1% and the placebo group's 12.7%. Furthermore, 53.6% of the Sotyktu treatment group achieved sPGA score of 0 or 1, higher than 32.1% in the apremilast group and 7.2% in the placebo group. In POETYK PSO-2 study, the Sotyktu treatment group had a PASI 75 response rate of 53.0% at week 16, which was significantly higher than the apremilast group's 39.8% and the placebo group's 9.4%. Additionally, 49.5% of the Sotyktu treatment group achieved sPGA score of 0 or 1, higher than the apremilast group's 33.9% and the placebo group's 8.6%. Sotyktu’s high response was maintained for up to 52 weeks. "Previously, patients who did not respond to or have had side effects when treated with conventional treatments, such as systemic therapy or light therapy, had biological agents as their only option. Sotyktu, which offers the convenience of once-daily oral administration, is expected to meet the unmet needs of psoriasis patients," Choe Yong-beom, President of the Korean Society for Psoriasis, said.
- Policy
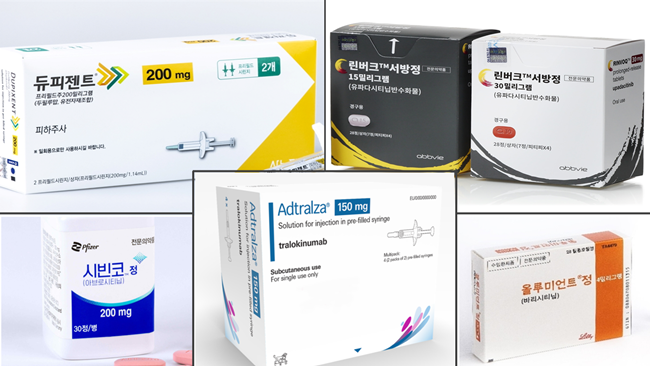
- Drug switching for atopic dermatitis measure submitted
- by Lee, Tak-Sun Nov 07, 2024 05:47am
- Pharmaceutical companies have submitted their measures for voluntary drug price reduction as part of financial allotment following expanded reimbursement. Discussions about allowing drug switching between biological agents and JAK inhibitors used to treat severe atopic dermatitis are speeding up. The Health Insurance Review and Assessment Service (HIRA) has already prepared the reimbursement criteria, and pharmaceutical companies have submitted their measures for voluntary drug price reduction as part of financial allotment following expanded reimbursement. Attention is drawn to whether it will pass the Drug Reimbursement Evaluation Committee (DREC) review, the final stage for reviewing reimbursement appropriateness. According to industry sources on November 6th, pharmaceutical companies Pfizer for Cibinqo and Lily for Olumiant submitted their financial impact analysis to the HIRA. The drug pricing reduction system for expanded scope of use requires the submission of financial impact analysis reports. The government excludes drug price reduction when the estimated additional claim amount following the expanded scope of use is below KRW 1.5 billion. The drug price reduction is applied for products over KRW 1.5 billion and below KRW 10 billion. In this case, pharmaceutical companies are required to submit a financial impact analysis, including measures to voluntarily lower prices, to the HIRA. In mid-October, the HIRA required each pharmaceutical company to submit financial impact analysis. Earlier this month, pharmaceutical companies have started to submit their reports. The issue of allowing drug switching for the treatment of severe atopic dermatitis was dealt with in the parliamentary audit held last month, so the health and welfare authority appears to be rushing the measure. The HIRA has been discussing with experts since September about whether to allow drug switching between biological agents and JAK inhibitors. The reimbursement criteria have been prepared, reflecting the latest evidence and clinical application. The remaining step is to minimize the financial impact following the expanded scope of use. Pharmaceutical companies' percentage of voluntary drug price reduction is the key. As pharmaceutical companies have submitted their measures for voluntary price reduction, the the Drug Reimbursement Evaluation Committee (DREC) review of the HIRA will make a final decision. The 11th DREC review is scheduled for July 2024. Attention is drawn to whether drug switching for severe atopic dermatitis will be considered for the agenda. When it passes the DREC review, the final reimbursement expansion will be decided through negotiation with the National Health Insurance Service (NHIS). Meanwhile, treatments for severe atopic dermatitis have been listed for reimbursement, starting with the biological agent, 'Dupixent Inj,' approved in January 2020. It was followed by the JAK inhibitors, Olumniant Tab, Linvoq ER Tab, and Cibinqo Tab. In May, another biological agent, Adtralza PFS, was listed for reimbursement. As a result, patients have more drug options. However, reimbursement and special cases do not cover drug switching between biological agents and JAK inhibitors, so it remains an issue. When clinical practices initially use high-cost drugs, and then the effects seen are not apparent, they may continue administering first-line treatments. Therefore, the medical community has been demanding the allowance of drug switching.
- Company
- Reimb discussions discontinued for NMOSD drug Uplizna
- by Eo, Yun-Ho Nov 07, 2024 05:47am
- Uplizna, a new drug for neuromyelitis optica spectrum disorder (NMOSD) that is administered twice a year, has failed to pass the final gateway to reimbursement in Korea. According to Dailypharm coverage, Mitsubishi Tanabe Pharma Korea’s pricing negotiations with the National Health Insurance Service (NHIS) for Uplizna (inebilizumab), a treatment used to treat adult patients with for neuromyelitis optica spectrum disorder (NMOSD) who are positive for anti-Aquaporin-4 (AQP4) antibodies, has been discontinued recently. The company had accepted the “below the evaluated amount” condition set by the Health Insurance Review and Assessment Service's Drug Reimbursement Evaluation Committee for the reimbursement of Uplizna (inebilizumab) in August but was unable to reach an agreement. NMOSD occurs when AQP4 autoantibodies, a disease-specific biomarker produced by B cells, bind to AQP4, a target antigen present on glial cells in the central nervous system, and activate the immune responses, causing nerve damage. Uplizna is an anti-CD19 human monoclonal antibody that selectively binds to CD19, a B cell-specific surface antigen, depleting B cells that produce AQP4 antibodies, thereby preventing disease relapse. The safety and efficacy of Uplizna were demonstrated in the N-MOmentum study, which evaluated the use of Uplizna monotherapy in 230 patients without the use of concomitant immunosuppressive agents. Study results showed that 89% of patients treated with Uplizna did not experience a relapse during 197 days of follow-up, resulting in a 77.3% reduction in the risk of relapse compared to placebo. Safety evaluations Uplizna also showed comparable rates of adverse events to the placebo group. Also, in an extension study, Uplizna continued to reduce the risk of relapse for at least 4 years, with an 87.7% relapse-free rate. In terms of long-term safety profile, Uplizna was generally well tolerated, with no increase in infection rates due to B-cell depletion. NMOSD is a serious autoimmune disease in which most patients experience persistent relapses and incomplete recovery, resulting in accumulated nerve damage that can cause vision loss, gait disturbances, and even death from respiratory failure.