- LOGIN
- MemberShip
- 2026-06-23 10:45:25
- Policy
- KDCA operates Public-Private Council for COVID-19 response
- by Kang, Hye-Kyung Aug 16, 2024 05:56am
- The Korea Disease Control and Prevention Agency (KDCA) announced that it will do its utmost to respond to COVID-19 by reorganizing its COVID-19 task force and forming a public-private council with experts. The agency expects the outbreak to continue until the end of August. On the 13th, the KCDA said, “The number of hospitalized COVID-19 patients began to increase from the end of June in the COVID-19 sampling surveillance. 861 were reported in the first week of August, approaching the peak of 875 hospitalized patients that was recorded in February this year. Considering the epidemic trend over the past 2 years, the number of COVID-19 patients is expected to increase until the end of August,” it said. The KCDA plans to expand its COVID-19 Task Force currently in operation. The response system, which was initially operated as 2 teams under 1 Task Force, will be expanded to 12 teams under five Units, with the head of the KCDA serving as the lead, to respond more quickly and thoroughly to epidemic investigation and analysis, overseas surveillance, and treatment supply and demand management. In addition, the KDCA explained that it had organized a public-private council with experts from the medical and academic fields related to COVID-19 to collect expert opinions, share the status of the COVID-19 outbreak with the medical community, and discuss countermeasures, and will hold regular meetings starting on the 14th. “This number of COVID-19-related hospitalizations has been increasing this summer, and it is likely to reach the scale of last year's summer epidemic, with 65% of confirmed cases occurring among the elderly aged 65 and older,” explained KDCA Commissioner Young-mee Jee. However, Commissioner Jee added, “Based on the analysis of domestic and international organizations on the KP.3 variant, which is currently the most common variant, the severity and fatality rates are not assessed to be significantly different from the previous Omicron variant, and the COVID-19 fatality rate in Korea in 2022-2023, which was after the Omicron outbreak, was less than 0.1%, especially for those under 50 years old. There is no need to be overly anxious.” “The most important thing to do for a healthy and safe summer is to take precautions to prevent infectious diseases, such as ventilating your home, washing your hands, and wearing a mask.”
- Company
- Twice-yearly Uplizna enters last stage to reimb in KOR
- by Eo, Yun-Ho Aug 16, 2024 05:56am
- Uplizna, a new drug for neuromyelitis optica spectrum disorder (NMOSD) that is administered twice a year, has entered the final gateway for insurance reimbursement in Korea. According to industry sources, Mitsubishi Tanabe Pharma Korea entered pricing negotiations with the National Health Insurance Service (NHIS) for Uplizna (inebilizumab), a treatment used to treat adult patients with neuromyelitis optica spectrum disorder (NMOSD) who are positive for anti-Aquaporin-4 (AQP4) antibodies. The company accepted the ‘below the evaluated amount’ condition set by the Health Insurance Review and Assessment Service's Drug Reimbursement Evaluation Committee for the reimbursement of Uplizna (inebilizumab) last month. The drug is administered at an initial 300 mg dose, followed by an additional 300 mg dose 2 weeks later, and then every 6 months thereafter from the date of the initial dose. NMOSD occurs when AQP4 autoantibodies, a disease-specific biomarker produced by B cells, bind to AQP4, a target antigen present on glial cells in the central nervous system, and activate the immune responses, causing nerve damage. Uplizna is an anti-CD19 human monoclonal antibody that selectively binds to CD19, a B cell-specific surface antigen, depleting B cells that produce AQP4 antibodies, thereby preventing disease relapse. The safety and efficacy of Uplizna were demonstrated in the N-MOmentum study, which evaluated the use of Uplizna monotherapy in 230 patients without the use of concomitant immunosuppressive agents. Study results showed that 89% of patients treated with Uplizna did not experience a relapse during 197 days of follow-up, resulting in a 77.3% reduction in the risk of relapse compared to placebo. Safety evaluations Uplizna also showed comparable rates of adverse events to the placebo group. Also, in an extension study, Uplizna continued to reduce the risk of relapse for at least 4 years, with an 87.7% relapse-free rate. In terms of long-term safety profile, Uplizna was generally well tolerated, with no increase in infection rates due to B-cell depletion. NMOSD is a serious autoimmune disease in which most patients experience persistent relapses and incomplete recovery, resulting in accumulated nerve damage that can cause vision loss, gait disturbances, and even death from respiratory failure.
- Company
- OraTicx signs a distribution deal with U.S.-based Stratum
- by Son, Hyung-Min Aug 16, 2024 05:56am
- OraTicx, a company specializing in developing oral probiotics, announced that it has signed an exclusive distribution agreement for the North American region with Stratum Nutrion (hereafter referred to as Stratum), a U.S.-based dietary supplement ingredient supplier. Through this agreement, OraTicx will exclusively distribute its proprietary oral probiotics 'oraCMU,' 'oraCMS1,' and 'oraCMU' in the North American market. Stratum, a company under ESM Technologies in the United States, provides natural ingredients based on scientific research. Through this partnership, OraTicx is expected to utilize Stratum's broad distribution network to accelerate the entry of its products into the North American market. OraTicx has 10 patents in South Korea and the United States. It has expertise in researching oral probiotics, completing 10 cases of human studies, and publishing 36 research articles. Notably, its oral probiotics strain, oraCMU, was developed in an OraTicx-affiliated research center. Through joint research with a research team at Seoul National University School of Dentistry, oraCMU's human study results demonstrating an improvement in halitosis were published in the SCIE international journal, 'Frontiers in Microbiology.' oraCMU demonstrated the quality and safety of the ingredient through FDA GRAS (Generally Recognized As Safe) registration. oraCMS1 has been confirmed to prevent plaque formation and exhibit antibacterial efficacy against respiratory pathogens. Recently, OraTicx has obtained a patent for its composition aimed at preventing and treating respiratory diseases. OraTicx has launched oral probiotics products using proprietary research. It is expanding its product line-up focused on oral probiotics, including Green Breath, Dentee, kids' dental probiotics, Implantics with twice the units, and Pet Biome for pets. OraTicX President Yoon Eun-seop said, "The current exclusive distribution agreement with Stratum is an important opportunity to introduce our innovative oral probiotics to North American customers." He added, "We aim to establish a new value in the North American dietary supplement market and become a leading global company contributing to people living heathy lives until the age of 100." Stratum President Micah Osborne said, "OraTicx's advanced oral probiotics will strengthen our portfolio even more." He added, "Through this partnership, we are delighted to provide various and effective healthcare solutions to North American customers."
- Policy
- KDCA 'COVID-19 drug supply will be stabilized within August'
- by Lee, Hye-Kyung Aug 16, 2024 05:55am
- The government has explained that pharmacies nationwide will have enough COVID-19 drugs by the last week of August. In the case of self-test kits, the government added that it plans to produce and supply more than 5 million kits within August to ensure no shortage. The Ministry of Health and Welfare (Minister Kyoo-hong Cho) held a joint meeting with the Korea Disease Control and Prevention Agency (KDCA) (Commissioner Young-mee Jee), the Office of the Government Policy Coordination (Minister Ki-sun Bang), the Ministry of Education (Minister Ju-ho Lee), the Ministry of Interior and Safety (Minister Sang-min Lee), Ministry of Food and Drug Safety (Director Yu-Kyoung Oh), and the National Fire Agency (Commissioner Seok-gon Heo) met together on the 14th at 4:30 p.m. to review Korea’s ‘COVID-19 epidemic trend and countermeasures’ in light of the recent increase in COVID-19 infections. The weekly usage of COVID-19 treatments as disclosed by the KDCA increased to exceed 42,000 doses in the 5th week of July, compared to the 1,272 doses used in the 4th week of July, exceeding the amount used during the summer season last year. To address the increased usage, the KDCA plans to supply additional drugs sequentially from this week, so that there will be enough drugs in all pharmacies that supply COVID-19 treatments from the last week of August (8.25~8.31). “We will endeavor to ensure smooth access to the treatment everywhere in the country by the last week of August,” the KDCA said. The Ministry of Health and Welfare plans to promptly review COVID-19 treatments that apply for reimbursement upon approval by the Ministry of Food and Drug Safety by hastening the reimbursement adequacy evaluation, NHIS negotiations, and Health Insurance Policy Review Committee deliberations. The MFDS is closely monitoring the supply of COVID-19 self-test kits and the entire production and distribution process. Domestic manufacturers have been scaling back production of self-test kits since COVID-19 turned endemic, but have recently expanded their production and supply to meet the rise in demand for COVID-19 test kits since late July. Korea’s COVID-19 self-test kit manufacturers own sufficient facilities, technology, and capacity to meet domestic demand, and are expected to produce and supply more than 5 million self-test kits within August. The MFDS added, “Korea’s COVID-19 self-test kit manufacturers have produced 40 million self-test kits per week in the past.” Meanwhile, the number of hospitalized COVID-19 patients has been increasing again since the end of June, and the number of hospitalized patients reached its peak this year (1,357, provisional) in the second week of August. While respiratory viruses are primarily prevalent during the winter season, COVID-19 has also been prevalent during the summer season (July-August) in 2022 and 2023, and given this trend, the number of COVID-19 patients is expected to continue to increase in the near future. According to the Ministry of Health and Welfare, the number of emergency room visits due to COVID-19 jumped from 2,240 in June to 11,627 in July. The MOHW said, “We plan to establish a cooperative system for hospitalization of COVID-19 patients by securing free beds, mainly in public hospitals that had been previously operated as dedicated COVID-19 hub hospitals. In order to ensure prompt treatment of COVID-19 patients in their local areas according to the pandemic situation, we plan to secure a list of COVID-19 treatment hospitals by local governments in cooperation with the Ministry of the Interior and Safety and local governments, and publish it through the Emergency Medical Services System.” The 2024-2025 COVID-19 vaccination plan will utilize a new vaccine (against the JN.1 strain) that is more effective against strains currently circulating in Korea and around the world. The government is currently in the process of licensing and approving the new vaccine so that it can be administered simultaneously with the flu vaccine in October, and details of the plan will be announced in September. “To respond to the COVID-19 pandemic, we will work with relevant ministries and organizations to secure beds so that COVID-19 patients can be treated promptly, and expedite the procurement of COVId-19 treatments and their reimbursement listing so that high-risk patients can stably receive treatment,” said MOHW Minister Kyoo-hong Cho. “The fatality rate of COVID-19 in Korea in 2023 (January-August), after the Omicron variant outbreak, was in the 0.05% range, and less than 0.01% for those under 50 years old, so there is no need to be overly anxious about the outbreak this summer,” said KDCA Commissioner Young-mee Jee. ”We will do our best to ensure that the additional supplies of COVID-19 drugs we secure from next week be supplied smoothly to patients for their convenient use.”

- Company
- K-Pharma speeds up marketing new drugs for bile duct cancer
- by Son, Hyung-Min Aug 16, 2024 05:55am
- Korean pharmaceutical companies are speeding up the development of new drugs for cholangiocarcinoma (also known as bile duct cancer). Handok's partnering company in the United States, Compass Therapeutics, has recently completed registering patients for the Phase 2/3 trials. Once the efficacy of the novel drug is confirmed, Handok plans to apply for approval in South Korea. According to pharmaceutical industry sources on August 16th, Compass Therapeutics has recently completed registering patients for the Phase 2/3 trials in the United States. The trials will evaluate the efficacy of HDB001A(CTX-009) for metastatic or recurrent cholangiocarcinoma and compare the efficacy of HDB001A in combination with paclitaxel to the paclitaxel monotherapy. HDB001A is a novel drug candidate developed by the Korean company ABL Bio. Handok has the domestic distribution rights, and Compass Therapeutics has the global distribution rights. This novel drug candidate is a bispecific antibody targeting Delta-like ligand 4 (Dll4) and vascular endothelial growth factor (VEGF), and it is known to form new blood vessels in the tumor microenvironment. Handok has confirmed the efficacy and safety of HDB001A in the Phase 2 trial conducted in South Korea. The clinical trials evaluated and compared HDB001A in combination with paclitaxel to paclitaxel monotherapy in 24 adult patients with advanced, metastatic, or recurrent cholangiocarcinoma who have received prior treatments. After a median follow-up of 12 months, patients treated with the combination therapy had an objective response rate (ORR) 37.5%, measuring the reduction in tumor sizes. The median overall survival (OS) was 12.5 months. The median duration of response (DOR) was 9.4 months, and the median progress-free period (PFS), duration without progression, was 9.4 months. However, the adverse events of HDB001A plus paclitaxel occurred at the highest rate. Treatment-related adverse events (TRAE) of all Grades, from 1 to 5, were reported in HDB001A plus paclitaxel combination therapy. 75% of patients had adverse reactions of over Grade 3. The most common adverse reactions over Grade 3 were neutropenia (50%), hypertension (16.7%), anemia (12.5%), and thrombocytopenia (8.3%). One case of Grade 5 pneumonia was reported. 25% of patients had confusion state, pulmonary embolism, and an increase in blood creatinine level. Once Handok confirms the efficacy and stable safety data from the U.S. clinical trial, it will apply for approval in South Korea. Confirming the potential of Keytruda combination therapy…enters the clinical trial Besides Handok, Genome & Company, GI Innovation, and SMT bio are developing novel drugs for cholangiocarcinoma. These companies are conducting clinical trials for Keytruda combination therapies. Korean pharmaceutical companies are conducting clinical trials for their candidate drugs in combination with various immune checkpoint inhibitors, including Keytruda.Keytruda was approved in April as a first-line treatment of cholangiocarcinoma in South Korea. Genome & Company, GI Innovation, and SMT bio plan to increase the possibility of commercializing their candidate products in combination with an efficacy-confirmed drug. Genome & Company is developing a new candidate product, microbiome-based immune checkpoint GEN-001, in combination with Keytruda for cholangiocarcinoma. Genome & Company aims to improve the efficacy of existing immune checkpoint inhibitors through combination therapy of 'GEN-001' and Keytruda. The company also plans to develop 'GENA-104,' an immune checkpoint inhibitor 'GENA-104' that suppresses novel target CNTN4, for patients who had not benefited from Keytruda targeting PD-L1/PD-1. The Phase 2 trial of GEN-001 for cholangiocarcinoma was approved for changes to the Investigational New Drug (IND) application in June. Genome & Company plans to evaluate the efficacy and safety of three-drug combination therapy, adding FOLFOX (fluorouracil, leucovorin, and oxaliplatin) to the existing GEN001 plus Keytruda combination therapy. Genome & Company confirmed the tolerability and safety of GEN-001 in the Phase 1 trial and determined the recommended dosage for the Phase 2 trial. The trial recently finalized patient treatments. SMT bio is conducting Phase 2b trials for allogenic Natural Killer cells (SMT-NK cells) in combination with Keytruda. The company is evaluating the efficacy of the combination therapy compared to the Keytruda monotherapy. Based on clinical results until now, the combination therapy had a PFS of 4.1 months, which is higher than 1.5 months of the Keytruda monotherapy. It showed improved ORR compared to monotherapy. Last year, the Ministry of Food and Drug Safety (MFDS) acknowledged the effectiveness of SMT-NK cells and approved the therapeutic use of under-trial pharmaceutical for treating patients with cholangiocarcinoma. GI Innovation is conducting a Phase 1 trial in the United States to evaluate the effectiveness of GI-101 in combination with Keytruda. The company aims to secure an indication for treating solid cancers, including cholangiocarcinoma. GI Innovation is also evaluating the marketing possibility of its candidate product in combination with a wide variety of immune checkpoint inhibitors, such as Imfinzi, in addition to Keytruda.
- Will Keytruda be reimb for its 17 indications in 2H?
- by Eo, Yun-Ho Aug 14, 2024 05:51am
- Whether any progress will be made in the immuno-oncology drug Keytruda’s reimbursement journey in the second half of the year is gaining attention. The application, which was first filed for 13 indications, has now grown to request coverage for 17 indications. In June last year, MSD Korea applied to extend the reimbursement coverage of its PD-1 inhibitor Keytruda (pembrolizumab) to 13 indications The 13 indications were: ▲ early-stage triple-negative breast cancer; ▲locally recurrent or metastatic triple-negative breast cancer, ▲metastatic or with unresectable, recurrent head and neck squamous cell carcinoma, ▲ locally advanced or metastatic esophageal or gastroesophageal junction (GEJ) carcinoma, ▲ adjuvant treatment of patients with renal cell carcinoma, ▲ non-muscle invasive bladder cancer, ▲ persistent, recurrent, or metastatic cervical cancer, ▲ advanced endometrial carcinoma, ▲ advanced endometrial carcinoma that is microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) ▲ unresectable or metastatic MSI-H or dMMR colorectal cancer ▲ metastatic MSI-H or dMMR small bowel cancer, ▲ metastatic MSI-H or dMMR ovarian cancer, and ▲ metastatic MSI-H or dMMR pancreatic cancer In December of the same year, the company submitted an application to include additional indications for MSI-H gastric cancer and MSI-H biliary tract cancer, and in April of the next year, 15 indications were submitted to the Health Insurance Reimbursement and Assessment Service’s Cancer Disease Review Committee for deliberation. At the time, the result was 'no reimbursement standard set', but the company also received a condition was the CDDC will rediscuss Keytruda’s reimbursement status if the company additionally submits a financial sharing plan. In other words, its review of the drug’s medical feasibility and medical necessity has been completed for 15 indications. In the first half of the year, the company also submitted applications to expand Keytruda’s reimbursement standards to HER2-positive (KN-811) and negative (KN-859) gastric cancer indications, which were approved in Korea in December last year and March this year, respectively. As a result, the company will be applying for the reimbursement of a total of 17 indications for Keytruda. As Keytruda is a Risk Sharing Agreement (RSA) drug, the company is required to go through a new drug-like evaluation process for each indication to receive reimbursement expansions. Indications approved through Phase III clinical trials must go through a pharmacoeconomic evaluation process to demonstrate cost-effectiveness, while indications approved based on Phase II trials must negotiate to receive an exemption from conducting the pharmacoeconomic evaluation. Therefore, it will be interesting to see if Keytruda, which is undergoing an unprecedented reimbursement expansion process, will be able to deliver results in the second half of the year. Meanwhile, the Padcev (enfortumab vedotin) + Keytruda combination was recently approved in Korea, heralding a paradigm shift in bladder cancer treatment. The combination of the two drugs in bladder cancer gained significant attention after the results of the Phase III EV-302/KEYNOTE-A39 study were presented at the European Society for Medical Oncology Annual Congress (ESMO Congress 2023) in October last year.

- Company
- LG Chem begins Phase 2 trials for infant hexavalent vaccine
- by Hwang, Byung-woo Aug 14, 2024 05:51am
- LG Chem LG Chem prepares to commence the Phase 2 clinical trial for its hexavalent vaccine LR20062. The company aims to domestically produce infant combination vaccines in South Korea. Since South Korea has so far relied entirely on imported combination vaccines, the company plans to provide a stable supply network to meet mid- to-long term demand for immunization. On August 13th, LG Chem announced it will conduct the Phase 2 trials for the hexavalent 'acellular Pertussis (aP)' vaccine, 'LR20062' overseas and prepare to enroll study participants. The successful completion of the Phase 1 trial and entry into the Phase 2 stage indicate progress towards commercializing the first domestically produced combination vaccine. 'LR20062' is a 6-in-1 vaccine used to prevent Diphtheria, Tetanus, Pertussis, Polio, Haemophilus influenzae type b (Hib), and Hepatitis B. Compared to vaccine conjugates in commonly used 5-in-1 vaccines and Hepatitis B vaccines, it reduced the number of doses by two doses (6 doses to 4 doses). The Phase 2 trial involving healthy adults demonstrated vaccine reactions from all study participants. Regarding immunogenicity endpoints, the serum protection and conversion rates were above 90%, similar to the control group, which was the existing hexavalent combination vaccine. In terms of safety, the new vaccine also demonstrated similarly favorable endpoints compared to the control group. The Phase 2 trial will enroll 300 infants aged two months and older, the target immunization age group in the real-world, as study participants and evaluate the safety and immunogenicity of 'LR20062' compared to existing hexavalent combination vaccines. LG Chem aims to develop domestically produced combination vaccines and contribute to meeting mid- to long-term demand through developing hexavalent combination vaccines. To this end, the company plans to invest about KRW 200 billion in R&D and facilities. Jeewong Son, Head of LG Chem Life Sciences Company businesses, said, "Establishing the production technology for all six antigens is a significant milestone and crucial for establishing domestically produced vaccines." He added, "We will expedite the commercialization of domestic combination vaccine to ensure that parents can safely and reliably vaccinate their children." Additionally, LG Chem is conducting the Phase 2 trial for a while cell Pertussis (wP) hexavalent vaccine, ‘LR19114,’ intending to enter the procurement market of international organizations like UNICEF.
- Policy
- Gov’t raises service fee for difficult surgeries
- by Lee, Tak-Sun Aug 14, 2024 05:51am
- The government is promoting a plan to completely reform the unbalanced structure of Korea’s fee-for-service system. For this, the government is reviewing whether to raise the compensation level for severe surgeries and reforming the relative value system and conversion index used to calculate medical service fees. Kyung-sil Jeong, Head of the Healthcare Reform Promotion Team at MOHW explained so at a briefing on the progress of Korea’s healthcare reform on the 13th. "Korea's reimbursement system is based on a 'fee-for-service system' that sets a unit price for every individual act of service," Jeong said, pointing out that "among the types of services, basic medical treatment, surgery, and treatment have lower fee levels, while testing, imaging, and functional type services have higher fee levels." The system has constantly been criticized as it incentivizes more tests than serious, high-risk surgeries. "We are promoting an overall reform to increase the compensation level in low-compensated areas and decrease the compensation level in high-compensated areas," Jeong said, adding, "First of all, we are considering raising the compensation level of about 1,000 severe surgeries, which are mainly performed at tertiary hospitals and general hospitals but have low fees." The government has established a 'Medical Cost Analysis Committee' within the Health Insurance Policy Review Committee to establish a system where medical fees can be quickly adjusted based on scientific evidence. The committee is also looking to reform the relative value system and conversion index, which are the basis of the fee-for-service system. "The committee has identified 6 priority areas that require concentrated investment: severe disease treatment, difficult essential medical treatment, emergency treatment, night and holiday treatment, pediatrics and childbirth, and vulnerable regions," Jeong said, adding, "In consideration of these priorities, we plan to more systemically introduce the public policy fee.” Furthermore, the government plans to transform the fee-for-service system into a "value-based payment system" to fundamentally improve the problem of how the current fee-for-service system increases the volume of procedures rather than improves the outcome of treatment. "The current system, which applies a uniform 15% premium to the reimbursement rate for services provided at tertiary general hospitals without distinguishing between severe and mild cases, will be converted to a system in which hospitals will be rewarded more for treating severe cases and less for treating mild cases," said Jeong, adding, "To this end, we will select and apply ‘appropriate disease groups' that are suited to the functions of each medical institution." Discussions are also underway to reform the management of non-reimbursed items and indemnity insurance. In July, the government formed a non-reimbursement and indemnity insurance subcommittee within the Specialized Committee for Essential Healthcare and Fair Compensation under the Special Committee on Healthcare Reform to discuss non-reimbursed management and indemnity insurance reform. During subcommittee discussions, it was suggested that non-reimbursed items with obvious excessive use concerns, such as manual therapy, cataract surgery with non-reimbursed lenses, and non-valve reconstruction, should be restricted, and that a management system should be established using the current positive listing system to set standard prices for non-reimbursed items that bring concerns over excessive use based on the non-reimbursed item status monitoring result and continuously manage the items through data analysis and re-evaluation. Regarding the reform of indemnity insurance, the role of the indemnity insurance as a complement to health insurance should be clarified and the system should be improved in line with this principle. During subcommittee discussions, an opinion was raised that the statutory coinsurance rate of the national health insurance should be adjusted to a reasonable level to minimize some of the negative impacts of indemnity insurance on the healthcare delivery system and access to healthcare. In particular, Jeong explained that there were suggestions that the scope of non-reimbursed coverage should be rationalized in conjunction with the government’s non-reimbursed item management measures to improve the issue of insufficient examination and follow-up due to the nature of indemnity insurance, where an agreement is made only between the insurer and the patient, and to reinforce the review and management system for proper use and supply of healthcare, "In order for the healthcare reform to succeed, we need to change the way people use healthcare, different to what they have been accustomed to," Jeong said, adding, "If you have relatively minor symptoms, please refrain from visiting emergency rooms of tertiary hospitals to make room for severe and urgent patients."
- Policy
- Coinsurance rate for chronic diseases reduced from 30%→20%
- by Lee, Hye-Kyung Aug 14, 2024 05:51am
- #1 The coinsurance rate for outpatient treatment at neighborhood clinics for chronic diseases such as hypertension and diabetes will be reduced by 10%, from 30% to 20%. The Ministry of Health and Welfare announced on the 13th that a partial amendment to the ‘Enforcement Decree of the National Health Insurance Act’ was approved at a cabinet meeting. The amendment includes the legal ground for a 10% reduction (from 30% to 20%) in the coinsurance rate for outpatient visits to clinics for chronic patients who meet the requirements notified by the Minister of Health and Welfare, such as applying for integrated management services for hypertension and diabetes and establishing and establishing and receiving review for customized management plans. The co-payment reduction system will be implemented from August 21st. The Ministry of Health and Welfare said that the amendment is expected to contribute to improving health by preventing complications, improving the healthcare delivery system, and encouraging rational use of healthcare by encouraging chronically ill patients to receive comprehensive and continuous management at neighborhood clinics. In addition to improving the coinsurance rate system for chronic diseases at the clinic level, the amendment also includes an exemption from reporting the total remuneration to the National Health Insurance Service if users submit a simplified withholding tax statement. Starting with the year-end settlement in 2025 (attributable to 2024), employers will be able to submit a simplified withholding tax statement (earned income) to the tax authorities, which will allow year-end settlement of insurance premiums through data linkage with the National Tax Service without reporting the total remuneration amount to the NHIS, which is expected to reduce the workload of employers. However, users should be aware that if they have not submitted a simplified withholding tax statement to the tax authorities, or if there is an error such as an omission of information on the simplified withholding tax statement, they will need to report the total remuneration amount of the previous year as they currently do. Also, to revitalize the monthly income adjustment system, the government has expanded the number of income brackets eligible for adjustment from 2 to 6 and made it possible to apply for adjustment not only if your income has decreased from the previous year, but also if it has increased. If a subscriber has temporarily earned more income than the previous year, he or she will be charged a higher premium next year, but from January 1, 2025, he or she will have the option to pay the higher premium in the year he or she earned the income through the monthly income adjustment process. The 2024 out-of-pocket maximum for the bottom 30% of the income bracket (income quintiles 1-3) will remain the same as last year to relieve the burden of out-of-pocket costs for medical expenses for lower-income subscribers. Also, to prevent social hospitalization in long-term care hospitals, the out-of-pocket maximum for nursing hospital stays of more than 120 days will be increased by the previous year's consumer price index (3.6%) for all quintiles (1st-10th), including those that fall in the bottom 30% of the income bracket. "The amendments to the Enforcement Decree are a follow-up to the 2nd Comprehensive National Health Insurance Plan announced in February," said Joong-gyu Lee, Director of the Bureau of Health Insurance Policy at the Ministry of Health and Welfare. "By unifying the year-end tax settlement report and expanding the scope of income monthly adjustment applications, these amendments will improve the convenience of insurance premium payments and contribute to easing the burden of medical expenses for chronically ill and low-income subscribers who receive comprehensive care."
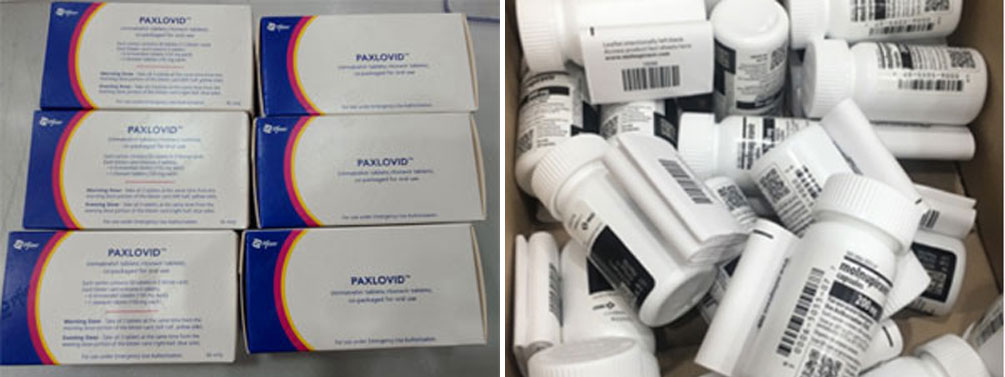
- Product
- Concerns arise about securing COVID-19 treatment shortages
- by Kang, Hye-Kyung Aug 14, 2024 05:51am
- COVID-19 is spreading due to the circulating KP.3 variant. We have not learned lessons from the previous spread of the Omicron variant. As demand for test kits surges, there are no remaining stocks at online pharmacies. Even test kits with an expiration date within the end of October are no longer available. Due to shortages of oral medicines, patients prescribed Paxlovid and Lagevrio visit one pharmacy after another or do not receive medicines. Now, the nation appears to be in chaos. Clinical practices are not aware of stock issues at the pharmacy, and pharmacies have no drugs. Local governments and the government are bombarded with inquiries. ◆"COVID-19 patients surged in a short period…prescriptions are made despite no drug availability"= The current situation is due to the surge in COVID-19 patients in a short period. After the announcement of the endemic, the number of patients showed a decreasing trend, but it has rapidly increased since July. Based on pharmacy data, an increased demand for COVID-19 self-test kits began on June 30th. According to the pharmacy data analytics service Care Insight (www.careinsight.co.kr), sales increased from ▲429 in June 30th-July 6th, ▲625 in July 7-13th, ▲1249 in July 14-20th, ▲2223 in July 21-27th, and ▲5850 in July 28th-August 3rd, doubling every week. A pharmacist 'A' in the metropolitan area said, "We have experienced an increase in COVID-19 patients since the end of July. We suspected it after seeing increasing demand for the test kits. The price for the test kits increased on demand from the end of July and the beginning of August, and now we are out of stock." The pharmacist added, "Even the remaining COVID-19 medicines sold out quickly." This pharmacy started receiving the distribution of COVID-19 medicines at the end of July. The pharmacist explained, "A public health center requested us to manage COVID-19 medicines tightly. Because we previously did not have that many number of patients, most pharmacies may not have a large quantity of stocks." The number of patients had been decreasing. When the medicines became charge-based, there were only two prescription cases within a month. The problem is that the number of patients is increasing but, there are not enough medicines. The Korea Disease Control and Prevention Agency (KDCA) expanded the distribution of medicines from once per week to twice per week, and the KDCA promised to distribute as much as pharmacies requested supplies. However, it is uncertain whether they can deliver this. This situation has arisen because we do not know how much COVID-19 medicines the KDCA has in stock. As the nation's demand for medcines keeps increasing, the supply requested from individual pharmacies is in short. Pharmacies that requested supplies from July 30th to August 5th have been rejected, and less than half of the stock is being delivered. A pharmacist 'B' in Seoul said, "We requested 96 drugs, but the quantity we received was 12, which we sold out of in 2 hours." The pharmacist added, "Other pharmacies within the same area are experiencing the same. There are cases where they requested but received none." As a result, the KDCA requested that prescriptions be made only for individuals over 60 years old with underlying diseases. The KDCA emphasized, "Please be advised to check with and prescribe COVID-19 medicines to high-risk patients with symptoms who are likely to progress to severe cases, thereby requiring oral medications, by the COVID-19 medicines guidelines." The KDCA requests clinical practices to prescribe only to people with ▲Tumors or hematological cancers ▲Congenital immunodeficiency disorders ▲'immuno-compromised individuals' like post-lung transplant patients ▲Diabetes ▲Hypertension ▲Cardiovascular diseases ▲Chronic kidney diseases ▲Chronic lung diseases ▲Body mass index (BMI) of 30kg/m2 or higher ▲Underlying diseases, such as neurodevelopmental disorder or mental illnesses. ◆Local authorities notify pharmacists to "come to local healthcare centers for drugs"'= Confusion occurs when the local government attempts to distribute medications during shortages. The KDCA has distributed additional quantities of medications through local public health centers. However, these quantities are insufficient to cover all designated pharmacies, leading to issues of fairness and equity between pharmacies. As of August 8th, it is understood that the distributed medication amounts to a supply of 15,000 people. In region 'D,' the government has announced that they will distribute and allocate medication in the same proportion, considering the orders placed this week and last week and the current usage levels at designated pharmacies. A local pharmacist said, "We received a message apologizing for a limited quantity due to evenly distributing additional medicines to all pharmacies." The pharmacist said, "However, we were told to come to local health centers to receive medicines since they cannot make deliveries to all pharmacies." In region 'E,' the distribution was confirmed to be handled on a first-come, first-served basis. A pharmacist from area 'E' stated, "On the afternoon of August 8th, the public health office official notified us via a social media group chat that only a tiny quantity would be available. They were accepting orders on a first-come, first-served basis. When I checked the message later, the stock had already been depleted." ◆KDCA says, "Temporary shortage, concerns are limited to particular areas"= The KDCA, the control tower for oral COVID-19 medications, shows an entirely different response. In response to criticism for rapid COVID-19 medicine usage and emptying Paxlovid inventory, the KDCA explained that "Particular regions may experience temporary shortages, but it is not true that stocks are running out." The KDCA said, "We thoroughly monitor real-time usage and inventory levels to prevent drug shortages. Collaborating with cities and provinces, we provide additional supply quantities to cities and provinces to respond to real-time demand in each region." They added, "If pharmacies are concerned about a shortage of medicines before the regular supply arrives, they can receive the supply management quantities from the local health centers." This response is entirely different from the situation at local pharmacies. The KDCA said, "However, the supply amount for individual pharmacies is determined based on actual usage, inventory levels, and the amount that can be distributed within the region. Therefore, it may not always match the requested quantities." The KDCA added, "We are working on additional purchases to protect high-risk individuals until stable supply within the general healthcare system is achieved."