- LOGIN
- MemberShip
- 2026-06-23 09:36:38
- Company
- Guidelines for new obesity drugs must be established
- by Moon, sung-ho Jul 26, 2024 05:46am
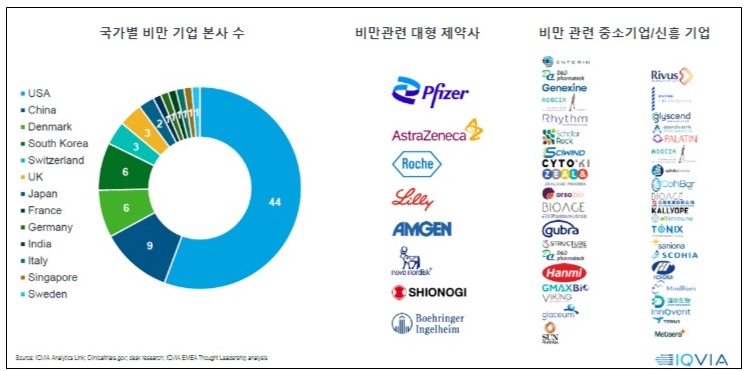
- Obesity is projected to impact around 2 billion individuals by 2035. Including overweight, about 4 billion individuals will be affected by obesity, making it a significant global healthcare issue. Following this trend, pharmaceutical and biotech industry is focusing on developing next-generation treatments for obesity. Glucagon-like peptide 1 (GLP-1) drugs, including Saxenda, Wegovy, and Zepbound, showed outstanding weight loss effects in clinical trials. As a result, companies are developing GLP-1-based therapeutics. The number of companies developing obesity drugs by country; big pharmas developing obesity drugs; small businesses and new companies developing obesity drugs. Recently, these drugs have been found to provide cardiovascular benefits and aid weight loss effects. As a result, their expanded use has been gathering attention in clinical settings. The analysis suggests that it's time to consider using these drugs in clinical settings in South Korea. According to drug market research company IQVIA on July 20th, there are 79 obesity drug pipelines worldwide, from preclinical to launched products. Pharmaceutical and biotech companies have developed over 148 products. GLP-1 drugs account for 39% of the pipelines. This suggests that pharmaceutical and biotech companies have begun developing drugs in this class as latecomers after witnessing the success of Saxenda, Wegovy, and Zepbound. 79 obesity drug pipelines worldwide, from preclinical to launched products, and 148 products from pharmaceutical and biotech companies have been developed. The analysis indicates that major companies that have begun developing obesity drugs employ two major development strategies. They either develop a drug as 'monotherapy' based on their differentiation strategy, or consider potential expansion for treating obesity, type 2 diabetes, cardiovascular diseases, and metabolic dysfunction-associated steatohepatitis (MASH) as part of their portfolio strategy. As part of differentiation therapy, monotherapy is being developed to achieve the ▲Highest weight loss rate, ▲Improved safety, and ▲Chronic disease management with oral formulation. For instance, Pfizer and Viking Therapeutics are developing candidate products. Under the portfolio strategy, the market leaders, such as Novo Nordisk (Wegovy) and Lily (Zepbound), and latecomers, aim to expand indications. Lately, the development of 'oral formulation drugs' has gained attention. These drugs are expected to shift the paradigm of a market dominated by injectables. Global companies, Pfizer and Viking Therapeutics, and domestic companies, including Ildong Pharmaceutical and D&D Pharmatech, have started to develop them. Novo Nordisk and Lily, the market-leading companies that already have injectables, have proprietary pipelines. However, Wegovy and Zepbound, which dominate the global market for obesity, have unresolved issue of weight loss rebound. IQVIA Korea's Marketing & Sales Director Kang-Bok Lee said, "Most obesity pipelines at the clinical stage are being developed as oral formulations." Lee added, "However, there are still discussions about oral obesity drugs. Along with the convenience, we must consider whether these drugs are suitable for chronic and maintenance management, and whether their cost and supply network outweigh any remaining issue." Lee added that "There are questions about whether it can have similar efficacy compared to injectables, as well as concerns about tolerability." Lee expressed optimism about oral drug development, saying, "Recently, Viking's oral drug, VK2735, had no clinically significant gastrointestinal side effects compared to placebo, with most being mild." Reflecting on the global trend for obesity drug development, new GLP-1 drugs, such as Wegovy and Zepbound, will likely be introduced to South Korea. According to IQVIA, the market for obesity drugs is rapidly growing after the launch of Wegovy. The worldwide market size in 2023 totaled US$11 billion (about KRW 15.3 trillion), driven by Wegovy. Wegovy contributed 72% of the total US$11 billion-worth market in 2023. In contrast, in South Korea, the release of Wegovy has been delayed due to an issue with 'securing stock' after obtaining marketing approval. As a result, Saxenda (Novo Nordisk) and Qsymia (Alvogen Korea) have taken 60% of the market share, dominating the market. Sources said that the release of Wegovy is set to be released in the Korean market, making it the ninth country globally. A professor from the Department of Endocrinology at an unnamed University Hospital, who is also an executive member of the Korean Society for the Study of Obesity, said, "Following Japan, China has also approved Wegovy. Since an official launch date has not yet been set, it is difficult to guarantee the timing of the release." He analyzed, "This appears to reflect the position of the domestic market within the global market." "Even if it is released, it seems that obesity drugs will be used entirely as non-reimbursable in the domestic market," He added. "While there has been some progress in recognizing obesity as a disease, reimbursement coverage, especially concerning domestic insurance finances, will not be easy." As a result, the pharmaceutical and biotech industries have proposed that, considering the potential of the drug market due to the rising prevalence of obesity, there is a need to accelerate discussions on disease recognition, enhancements in social awareness, and the establishment of clinical practice guidelines and insurance. At the same time, domestic pharmaceutical and biotech companies developing treatments may need to focus on differentiation strategies from competing products, improving efficacy such as preventing weight regain after discontinuation and establishing a stable supply chain to meet global demand. IQVIA Korea's Director Kang-Bok Lee said, "In the past two years, global spending on obesity has increased rapidly with new drugs, and by 2030, more than 15 new items are expected to enter the market, making the next-generation obesity treatment market much more competitive." Lee also said, "Improving educational programs for healthcare professionals to raise awareness of obesity treatment and integrating it into chronic disease management would be an ideal approach." Lee also added, "Currently, even though obesity treatments are approved by the Ministry of Food and Drug Safety (MFDS), there are no cases where these drugs are covered by reimbursement. In the U.S., Medicare (Part D) will now cover Wegovy for some patients with a history of heart disease, as announced by the Centers for Medicare & Medicaid Services (CMS)." Lee added, "In the future, obesity drugs will be divided into reimbursed and out-of-pocket markets, so it is necessary to consider establishing reimbursement criteria for patients with severe obesity or accompanying diseases. We must develop and distribute comprehensive obesity treatment guidelines through collaboration with the medical community and organizations."
- Company
- Antibiotic prescriptions had surged with the pandemic
- by Kim, Jin-Gu Jul 25, 2024 05:51am
- The amount of outpatient antibiotic prescriptions in Korea had changed dramatically during the COVID-19 pandemic. In 2020 and 2021, early stages of the pandemic, antibiotic prescriptions dropped sharply but then surged in 2022. In contrast, the proportion of cephalosporin and quinolone antibiotic prescriptions, which are more powerful than other antibiotics, increased in 2020 and 2021 and then decreased in 2022. The Ministry of Health and Welfare released the "Healthcare Quality Statistics" that contained the above findings on the 24th. The MOHW releases this data annually for comparative statistics among OECD countries. One of the statistical items examines outpatient antibiotic prescriptions in primary care clinics. According to the statistics, in 2022, outpatient prescriptions of local antibiotics, not systemic antibiotics amounted to 21.3DDD per 1000 inhabitants in primary healthcare centers in Korea. Defined Daily Dosage (DDD) is a unit used to measure drug consumption and refers to the average maintenance dose that an adult weighing 70 kg should take per day. Total outpatient antibiotics prescribed for systemic use (Source: Healthcare Quality Statistics 2022) The number of antibiotic prescriptions in Korea has been steadily decreasing since 2016 when it reached 26.9 DDD. Especially during the pandemic, the number dropped below 20.0 DDD in 2020-2021. It is analyzed that as people refrained from outdoor activities due to social distancing measures, respiratory infections decreased, reducing antibiotic prescriptions. In 2022, when the social distancing measures were eased, antibiotic prescriptions made a rebound. From 16.0 DDDs in 2021 to 21.3 DDDs in 2022, the amount increased 33% in 1 year. In contrast, prescriptions for cephalosporin and quinolone antibiotics, which are broader and more potent than other antibiotics, increased significantly early in the pandemic and then decreased near the endemic. Among all outpatient antibiotic prescriptions, the percentage of cephalosporin and quinolone antibiotics increased from 39.5% in 2019 to 43.6% in 2020, then to 44.8% in 2021. In 2022, their share decreased slightly to 43.1%. Percentage of cephalosporin and quinolone antibiotics prescribed (Healthcare Quality Statistics 2022) The prescription and use of antibiotics require management due to resistance issues and is considered one of the important areas to monitor through national antimicrobial resistance management policies. The OECD Healthcare Quality Statistics also includes two indicators related to antibiotics: the total number of outpatient antibiotics prescribed for systemic use and the proportion of cephalosporin/quinolone antibiotics prescribed. The MOHW explained, "The total volume of outpatient antibiotic prescriptions decreased by 34% in 10 years since 2011 due to the strengthening of antibiotic management policies and increased public awareness. However, the proportion of cephalosporin/quinolone antibiotics prescribed for systemic use has continued to increase over the decade and remains high at 43% as of 2022."
- Policy
- Samjin and Korea Pharma’s first generics are reimb in KOR
- by Lee, Tak-Sun Jul 25, 2024 05:51am
- Samjin Pharmaceutical and Korea Pharma have launched first generics in Korea. Samjin Pharmaceutical will launch the first generic of Sanofi's atrial fibrillation drug Multaq Tab, while Korea Pharma will launch the first generic of Pfizer's antidepressant Pristiq ER Tab. According to industry sources, Samjin Pharmaceutical's ‘Samjin Dron Tab as (dronedarone)’ will be listed at KRW 808 per tablet in Korea. The drug is indicated for the management of atrial fibrillation (AF) in patients in sinus rhythm with a history of paroxysmal or persistent AF to reduce the risk of hospitalization. Adults may take 1 tablet (dronedarone 400 mg) twice daily with breakfast and dinner. The original generic version of dronedarone is Sanofi's Multaq Tab. It was approved in Korea in February 2010. The product patent for Multaq expired in 2022. According to UBIST, its last year's outpatient prescriptions amounted to KRW 10.9 billion, exceeding KRW 10 billion for the first time last year. As Samjin already owns a high market share in the field of cardiovascular disease withPlatless (clopidogrel) among others, Samjin Dron Tab is expected to settle in the market in the short term. In particular, sales growth will further accelerate as there are no generic competitors in the market. Korea Pharma will launch the first generic version of Pfizer's Pristiq 100mg. Pristiq’s generics are already available in the market. In 2020, 4 domestic pharmaceutical companies entered the market with salt-modified drugs that evaded the patents. Therefore, the currently available follow-on drugs have different ingredients from the original product. Whereas Pfizer's Pristiq contains desvenlafaxine succinate monohydrate, the salt-modified drugs contain desvenlafaxine benzoate or desvenlafaxine. Korea Pharma’s ‘Pharma Desvenlafaxine ER Tab. 100mg’ is a first generic that uses the same salt as the original. Desvenlafaxine is a serotonin-norepinephrine reuptake inhibitor (SNRI), which is characterized by its low risk of drug interactions and a low risk of side effects such as hypertension and sexual dysfunction. Last year, Pristiq generated sales of KRW 7.2 billion according to IQVIA. Its sales have been declining with newer generics gradually gaining influence in the market. A representative from Korea Pharma said, "Pharma Desvenlafaxine ER Tab is a competitive product that was first approved with the same salt formulation as the original Pristiq ER Tab. We plan to quickly land and expand our market share through active sales activities." Samjin Pharmaceutical and Hankook Pharma's products received an insurance price of 59.5% of the original’s insurance ceiling price instead of 53.55% because the two companies have listed the first and only first generics. As a result, Samjin Dron Tab was listed at KRW 808 and Pharma Desvenlafaxine ER Tab at KRW 742 in Korea. Their original versions, Multaq Tab and Pristiq ER Tab are priced at KRW 1,357 and KRW 1,247, respectively.

- Company
- Meningitis B vaccine Bexsero is released in Korea
- by Moon, sung-ho Jul 25, 2024 05:51am
- Competition in the 'meningococcal' vaccine market, which is mainly vaccinated in pediatric clinics, has recently been reignited. Although its domestic market is worth less than KRW 10 billion, the emergence of next-generation vaccines is expected to spark new competition among multinational pharmaceutical companies. #This is because GSK, the market leader, has launched Bexsero (meningococcal serogroup B vaccine), a next-generation vaccine. Professor Hyunmi Kang (Department of Pediatrics, St. Mary's Hospital, Seoul) explained the clinical implications of the introduction of Bexsero at an event held by GSK on the 16th. Meningococcal meningitis is a statutory Class 2 infectious disease with a fatality rate of approximately 10-14%. It affects 500,000 patients worldwide each year. The main symptoms include headache, fever, neck stiffness, vomiting, and decreased consciousness, and is often accompanied by petechiae or purpura fulminans. 11 to 19% of recovered patients may suffer from sequelae such as hearing loss, cognitive impairment, and neurological disorders, making it an important infection to prevent. In particular, as meningococcal disease is transmitted person-to-person by respiratory droplets or secretions, vaccination is recommended for those who are about to enter a group setting. For example, new recruits and college students who will be living in dormitories may want to consider meningococcal vaccination. Other recommended populations for meningococcal vaccination include travelers and residents of meningococcal endemic areas, such as Africa, and pilgrimage travelers to Mecca, Saudi Arabia. Typical serogroups of meningococci that cause invasive meningococcal infections in humans include A, B, C, W, X, and Y. The most predominant meningococcal serogroup in Korea among these is serogroup B. From 2010 to 2016, the proportion of Meningitis B cases identified in Korea was 28%, but from 2017 to 2020, the rate rose significantly to 78%. In this scene, GSK launched Bexsero, a vaccine that prevents meningococcal serogroup B, in the Korean market 2 years after its approval in 2022. Professor Kang assessed that Bexsero can play a significant role in addressing the unmet need in the field as it prevents meningococcal serogroup B. "Globally, meningococcal infections are most prevalent in infants under one year of age compared to other age groups,” explained Professor Kang. “It causes bacterial meningitis and sepsis, and one to two out of 10 survivors also experience brain damage, hearing loss, and limb loss.” "The prevalence of meningococcal serogroups varies across countries and time periods, so it is not easy to predict. In Korea, serogroup B meningococcal infection cases have increased in recent years, increasing the need for its prevention.” In Korea, GSK is leading the meningococcal vaccine market. The vaccines, which are non-reimbursed, cost KRW 150,000. The first quadrivalent meningococcal vaccine in Korea, GSK’s Menveo has been dominating the market with sales of KRW 5.2 billion based on IQVIA last year. Sanofi's Menactra is also available, but the vaccine only generated KRW 500 million in sales during the same period. In addition, Sanofi received domestic approval for Menquadfi Inj (meningococcal (A, C, Y, W) tetanus toxoid-conjugate vaccine) earlier this year. "In countries such as the United Kingdom, Portugal, and Canada, the importance of preventing disease through immunization has been emphasized due to the high prevalence of meningococcal B," said Joon Bang, Director of Medical Affairs at GSK Korea. "The predominance of meningococcal B in Korea has made it necessary for us to introduce a vaccine to prevent infections caused by meningococcal B.” "We are pleased to be able to contribute to the prevention of meningococcal disease caused by serogroup B, which accounts for a high proportion of meningococcal disease in Korea with the launch of Bexsero. Together with Menveo, the company now owns a vaccine portfolio that can protect against a wide range of serogroups."

- Company
- 'Padcev+Keytruda' combination therapy is set to land in KOR
- by Eo, Yun-Ho Jul 25, 2024 05:51am
- Product photo of Padcev.The combination therapy of 'Padcev+Keytruda,' which is expected to bring a paradigm shift to bladder cancer treatment, will soon land in South Korea. The Ministry of Food and Drug Safety (MFDS) is reviewing the expansion of indication for Astellas Korea's Padcev (enfortumab), an antibody-drug conjugate (ADC), in combination with Keytruda (pembrolizumab), a PD-1 inhibitor that is used in immunotherapy for cancer, as a first-line treatment of urothelial cancer locally advanced or metastatic urothelial carcinoma (la/mUC). The official approval is expected soon. The combination therapy of these drugs for urothelial cancer gained attention after the presentation of its Phase 3 EV-302/KEYNOTE-A39 study results at the 2023 congress of the European Society for Medical Oncology (ESMO Congress 2023), held in October last year, and received a standing ovation. Based on the clinical results, patients treated with Padcev combination had a median progression-free survival (PFS) of 12.5 months, a primary endpoint of the study, indicating a significant improvement compared to the 6.3 months in patients treated with chemotherapy for cancer. Another primary endpoint of the study was median overall survival (OS). The patients had a median OS of 31.5 months, a twofold extension compared to the placebo. Cisplantin-eligible patients treated with Padcev combination had an OS of 31.5 months compared to 18.4 months in placebo-treated group, reducing the risk of death to 47%. Cisplantin-ineligible patients had not reached the median value, whereas the placebo-treated group had an OS of 12.7 months, reducing the risk of death to 57%. In clinical settings, 'Padcev+Keytruda' combination therapy is being considered as a replacement for a first-line treatment GemCis therapy, which has been used as the standard therapy for 30 years. Meanwhile, Padcev was approved in South Korea in March 2023 as a monotherapy for patients with locally advanced or metastatic urothelial cancer who had previous experience with platinum-containing chemotherapy or PD-1 or PD-L1 inhibitors. Astellas has applied for the inclusion of this therapy in reimbursement listing, and it has passed the Cancer Disease Review Committee of the Health Insurance Review and Assessment Service (HIRA).
- Opinion
- [Reporter's View] Keeping up with the Chinese bio industry
- by Son, Hyung-Min Jul 24, 2024 05:51am
- The research and development (R&D) capabilities of Chinese pharmaceutical companies are going from strength to strength year after year. Last year, the immuno-oncology drug Loqtorzi, developed by Junshi Biosciences, was approved in the United States. It became the first Chinese immuno-oncology drug to be approved by the U.S. Food and Drug Administration (FDA). While Chinese anti-PD-L1/PD-1-targeted immuno-oncology drugs have been approved within China, this is the first time they have crossed the FDA approval threshold. This year, BeiGene’s immuno-oncology drug Tevimbra was approved by the FDA. Another Chinese drugmaker, Innovent, is also seeking FDA approval for its own immuno-oncology drug sintilimab in partnership with Eli Lilly. The domestic pharma and biotech industry has also launched a number of immuno-oncology drugs, but most of them are still in the pre-Phase II stage. This is in contrast to Chinese companies, which have completed Phase III trials and are closer to global commercialization. China is leading the way in global commercialization not only of immuno-oncology drugs but also of next-generation therapies such as targeted anti-cancer drugs, gene therapies, and nucleic acid therapeutics. On the other hand, Korea has not been able to make a significant impression in the global market outside of biosimilars. In fact, the number of global new drug approvals by Korean pharmaceutical companies is incomparable, far less than that of Chinese pharmaceutical companies. The number of global new drug approvals by Chinese pharmaceutical companies in countries other than the US has also been steadily increasing, receiving approvals for 44 new drugs in 2020, 40 in 2022, and 14 last year. In the United States, 11 Chinese-made drugs have been approved in the past 3 years. In the case of Korean pharmaceutical companies, only 8 new drugs have been approved so far. In terms of clinical trials, China's share grew by 6.1% from 2019 to 2023, while Korea's share grew by only 0.3% during the same period. Government support has played a major role in raising the Chinese pharmaceutical industry's R&D capabilities. The Chinese government has deisgnated the pharma-bio industry as its economic growth engine and has shortened the approval time for drugs by innovating the CFDA’s review system. Examples include priority review drugs, expanding the number of Centers for Drug Evaluation (CDEs), and accepting overseas clinical data. As a result, China's clinical and new drug review time has been reduced by about one-third compared to before 2015. The expansion of China’s insurance reimbursement that increased the number of drugs on the reimbursement list has also worked in favor of Chinese pharmaceutical companies. The Chinese government continues to expand insurance reimbursement coverage of new drugs, and the revenues are being channeled into R&D investments. Korea, on the other hand, has been less successful in supporting R&D. The Yoon Suk-Yeol's government pledged to support bio R&D from the beginning of his administration, pledging to ‘establish a pharma-bio control tower’ to integrate, foster, and support the pharma-bio industry. However, to date, cooperation between the government and the industry has not been active, as the control tower, the Biohealth Innovation Committee, has not been fully operationalized, and regulations restricting the pharmaceutical industry, such as the drug price reduction system and reimbursement adequacy reevaluations, continue to increase. In order to create new global drugs, Korea needs to establish specific regulations and support systems for innovative drug development. It is illogical to expect good drugs to be made while investing less. Only more investment can lay the foundation for the development and launch of new global medicines. The government has budgeted KRW 2.9 trillion for fundamental R&D next year, which is the largest amount ever allocated. It is this reporter’s hope that this will spur the biotech industry to invest in challenging research areas and develop innovative new drugs.

- Policy
- Gvt commences 'Machine Learning AI Project' for new drug dev
- by Lee, Jeong-Hwan Jul 24, 2024 05:51am
- The government will actively pursue a project for faster new drug discovery. It will fund KRW 34.8 billion over 5 years, from 2024 to 2028, and conduct a project utilizing federated learning of new drug discovery data owned by pharmaceutical companies, university hospitals, research institutes, and corporations. The Ministry of Health and Welfare (MOHW) and Ministry of Science and ICT (MSIT) announced on July 23rd that 26 projects have been selected for the 'Machine Learning Ledger Orchestration for Drug Discovery (K-MELLODY)' project. The MOHW and MSIT are jointly promoting the MELLODDY project, which aims to reduce the cost and time for new drug discovery using a federated learning-based AI model that can safely protect data while exploring. Federated learning is a decentralized learning approach that enables machine learning to be trained internally on data from multiple locations, such as private and institutes, without directly sharing. Analyzed data are sent to a central server. The government has commenced this project and designated the Korea Pharmaceutical and Bio-Pharma Manufacturers Association (KPBMA) to run the project. The project group has selected 26 subprojects and research institutes for each project through a public survey and evaluation. The selected subprojects are categorized into three fields: ▲Establishing Federated Drug Discovery (FDD) platform, ▲Utilizing new drug discovery data and quality management, and ▲Developing AI solution. First, for 'Establishing Federated Drug Discovery (FDD) platform,' project has been selected to establish FDD platform to enable machine learning safely without sharing data between institutes and keeping privacy. As a supervision agency, Evident Inc. has been chosen. Also, 20 institutes have been selected as supervision agencies for AI-driven drug discovery. They will share new data from the fields of drug metabolism·toxicity testing and pharmacoepidemiology. Pharmaceutical companies, universities·hospitals, research centers, and corporations are included. Eight pharmaceutical companies have been selected, including Daewoong Pharmaceutical, Dong-wha Pharm, Samjin Pharm, Yuhan Pharm, Jeil Pharm, Hanmi Pharm, Huons, and JW Pharmaceutical. Universities·hospitals include Gachon University, Catholic University of Korea, Kyungpook National University, Seoul National University, and Seoul National University Hospital. Participating research centers·foundations include Daegu Gyeongbuk Advanced Medical Industry Promotion Foundation, KRIBB, Institut Pasteur Korea, and KRICT. Selected corporations include CIMPLRX and Apace. For 'developing AI solution,' five institutes, including GIST, Mogam Institute for Biomedical Research, AIGEN Sciences, Chonbuk National University Industrial Cooperation Foundation, and KAIST. They will develop the 'Federated ADMET Model (FAM)' to aid in drug candidate discovery, using experimental data from all stages of new drug discovery. The MOHW and MSIT will allocate KRW 34.8 billion to these selected projects over a five year period, from 2024 to 2028. They anticipate establishing an AI-driven new drug discovery infrastructure by collaborating with the government, pharmaceutical companies, research centers, and universities. "By developing machine learning with a federated learning approach that safely shares and uses data, previously challenging large-scale data analysis and use will be enabled, accelerating drug discovery," Ko Hyeongu, MOHW's Advanced Healthcare Support Officer, said. Ko said, "We will strengthen the data usage system and support AI‧Data R&D. We will also strive to lead future healthcare and pharmaceutical innovation and promote citizen's welfare." "The federated learning approach will enable the safe utilization of high-quality drug discovery data accumulated from multiple institutes to develop an AI-driven drug discovery platform and machine-learning solution," Kwon Hyeonjun, MSIT's R&D Policy Director, said. "We will support the R&D of digital bio-health businesses, which converges digital technology, and creating value so that South Korea emerges as a hub for cutting-edge biotechnology."
- Policy
- Roche’s Phesgo is applied RSA for reimb in Korea
- by Lee, Tak-Sun Jul 24, 2024 05:51am
- Korea’s insurance authorities are introducing various measures to reduce drug costs. The government is applying risk-sharing agreements to precalculation drugs and requiring submission of follow-up data for drugs that are exempt from sumitting pharmacoeconomic evaluation data. The drugs subject to the authorities’ measures are Phesgo SC Inj (pertuzumab/trastuzumab) and Ilaris Solution (canakinumab). Phesgo is a biobetter that was developed as a fixed-dose subcutaneous injection formulation of the intravenous injected Herceptin and Perjeta to improve dosing convenience and reduce treatment time for breast cancer patients. Since it is approved as an improved biologic, its insurance price was automatically set at 110% of the upper limit of its targeted products. As a result, Phesgo SC Inj 600/600mg will be listed at KRW 3,490,410, and Phesgo SC Inj 1200/600mg at KRW 5,914,418. According to IQVIA, sales of Herceptin and Perjeta in Korea amounted to KRW 56.5 billion and KRW 111.3 billion, respectively, last year. As an improved version of the two drugs, Phesgo is also expected to record high sales. However, its impact on insurance finances is also expected to be significant. This is why the insurance authorities decided to apply RSA to Phesgo and save insurance finances. The decision was influenced by the fact that Phesgo’s development target, Perjeta, is currently reimbursed through RSA. Like Perjeta, Phesgo is applied the refund-type RSA, under which the company refunds a certain percentage of the claims. "Considering that Phesgo’s target product is an RSA drug, we decided to apply RSA to Phesgo through negotiations with the National Health Insurance Service," explained the insurance authorities. “The product has improved the administration route of the target product, which is convenient for patients to administer, and we expect it to bring financial savings when it replaces the target product." Phesgo passed the Health Insurance Review and Assessment Service’s Cancer Disease Review Committee in August last year, but it took more than a year for it to be listed for reimbursement, the delay which can be explained by the cost-saving measure. Ilaris, which is being reimbursed after 9 years of approval in Korea, also has a number of safeguards in place to reduce costs. It is applied to the refund-type and expenditure cap-type RSA, and conditional follow-up is also required as a PE exemption drug. Its list price is KRW 11,029,469 Ilaris is a rare disease drug for which there are no alternatives and is being publicly reimbursed in over 3 of the A8 countries, allowing the drug to omit submission of pharmacoeconomic evaluation data. However, HIRA’s panel determined that the drug needs to be followed up in the future, including reevaluation of clinical effectiveness and cost-effectiveness, given the uncertainty of its improvement in clinical utility and high cost. Therefore, HIRA imposed a condition on the pharmaceutical company to conduct a prospective clinical study (observation period of 2 years for each patient and submission of observation data and results in 1-year increments) and submit clinical utility and cost-effectiveness data at the end of the RSA term. For example, the pharmaceutical company is required to conduct relevant prospective clinical studies, in an objective, cross-sectional survey format, with at least 2 years of observation per patient, and submit observations and results on a yearly basis. For the NOMID (neonatal onset multisystemic inflammatory disease)/CINCA (chronic infantile neurocutaneous joint syndrome) indication, the company needs to submit efficacy comparison data with existing treatments and cost-effectiveness data at the end of the RSA term. In addition, details such as plans to conduct prospective clinical studies for follow-up management and measures to be taken in case of failure to conduct such studies are required in the RSA. "In the future, drugs that are exempted from economic evaluation, but whose clinical usefulness is unclear, will be thoroughly reevaluated through follow-up management measures," explained a HIRA official.
- Company
- Samsung Bioepis’ Soliris biosimilar is approved in the U.S.
- by Chon, Seung-Hyun Jul 24, 2024 05:51am
- View of the Samsung Bioepis building [데일리팜=천승현 기자] 삼성바이오에피스는 미국 식품의약품국(FDA)으로부터 희귀질환치료제 ‘에피스클리’의 품목허가를 획득했다고 23일 밝혔다. 에피스클리는 미국 알렉시온이 개발한 솔리리스의 바이오시밀러 제품이다. 에피스클리는 발작성 야간 혈색소뇨증, 비정형 용혈성 요독 증후군의 치료제로 FDA 승인을 받았다. 삼성바이오에피스는 2019년 7월부터 2021년 10월까지 발작성 야간 혈색소뇨증 환자들을 대상으로 임상 3상을 통해 에피스클리와 오리지널 의약품 간 비교 연구를 수행했다. 유관 학술대회 발표를 통해 임상의학적 동등성을 입증했다. 삼성바이오에피스는 지난해 7월 에피스클리를 유럽에 출시했고 독일, 이탈리아 솔리리스 바이오시밀러 시장 점유율 1위 등의 성과를 내고 있다. 국내에서는 지난 4월부터 삼성바이오에피스가 직접 에피스클리를 판매하고 있다. 삼성바이오에피스는 오리지널 의약품의 약가 대비 절반 수준으로 솔리리스 바이오시밀러를 출시했다. 삼성바이오에피스는 이번 허가로 미국 시장에서 총 8개 바이오시밀러 제품을 승인받았다. 미국에서도 자가면역질환 치료제, 항암제, 안과질환 치료제에 이어 혈액·신장질환 치료제 분야까지 치료 영역을 확대했다. 삼성바이오에피스 고한승 사장은 “미국에서도 희귀질환 치료제를 승인받아 글로벌 수준의 R&D 역량을 인정받았다”며 “에피스클리는 바이오시밀러의 사회적 가치를 극대화할 수 있는 제품으로서 세계 최대 의약품 시장인 미국에서도 그 가치를 실현하기 위해 지속 노력하겠다”고 전했다.

- Company
- Reimb applied for new HIV drug 'Vocabria+Rekambys'
- by Eo, Yun-Ho Jul 24, 2024 05:50am
- Long-acting HIV treatment 'Vocabria+Rekambys' combination therapy aims to be listed for insurance reimbursement after receiving approval in South Korea two years ago. Industry sources said that GSK Korea and Janssen Korea have applied for reimbursement for the combination therapy of Vocabria (cabotegravir) and Rekambys (rilpivirine), which each company owns. GSK will be responsible for the overall reimbursement process. In February 2022, two drugs were approved by the Ministry of Food and Drug Safety (MFDS) as a combination therapy for the treatment of adult patients with HIV-1 infection who are virologically suppressed with no history of treatment failure and with no known or suspected drug tolerance to either cabotegravir or rilpivirine. In South Korea, Vocabria +Rekambys combination therapy was approved as injection therapy with intervals of monthly or bimonthly administration. The advantage of this combination therapy is its convenience. Previously, patients had to take a tablet formulation drug once daily for conventional HIV treatments. However, with the marketing approval of these two injectables, the treatment frequency has been reduced to monthly or bimonthly as an intramuscular injection, resulting in high patient satisfaction and reduced patient burden. These two drugs were initially developed as oral formulations and later developed as injectables. As a long-acting injection therapy, these drugs cannot cure HIV injection but target white blood cells, helping to lower and maintain AIDS virus replication. The combination therapy was approved in Europe in December 2020 after a clinical trial demonstrated its efficacy and safety in a patient group treated once every 4 weeks or once every 8 weeks. The most common adverse reactions observed in a group treated with Vocabria+Rekambys combination therapy include injection site reaction, headache, fever, nausea, fatigue, general malaise, and muscle aches. Consequently, it remains to be seen whether the government agency will approve the combination therapy of these two drugs for their advantage in convenience and whether the therapy will later become listed for reimbursement.