- LOGIN
- MemberShip
- 2026-06-23 10:45:24
- Company
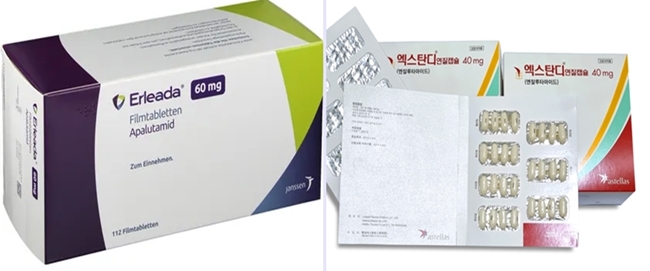
- 'Xtandi and Erleada' compete in the prostate cancer market
- by Hwang, Byung-woo Aug 14, 2024 05:51am
- The market for ARTA treatment used to treat prostate cancer has been dominated by Xtandi (enzalutamide). As prescription sales of Erleada (apalutamide) rise, the market is shifting. Astellas Pharma's Xtandi has increased its competitiveness with its indications, Janssen has started to face competition by expanding its portfolio, including Erleada, Zytiga, and Akeega. (From left) product photos of Erleada and Xtandi According to the pharmaceutical market research firm UBIST, Xtandi generated the highest outpatient prescription sales in this year's first half, KRW 14.1 billion. Analysis suggests that increased outpatient prescription sales are due to expanded reimbursement. In August 2022, Xtandi was selectively reimbursed for the treatment of patients with advanced prostate cancer accompanied by distant metastasis when used in combination with androgen deprivation therapy (ADT). Since November of last year, reimbursement has been made regardless of using other androgen synthesis inhibitors. However, the drug price was reduced from KRW 20,882 to KRW 14,170 due to the expanded reimbursement range, affecting prescription sales. Xtandi's prescription sales for this year's Q3 was KRW 8.1 billion, then reduced to KRW 6.6 billion in Q4. In November last year, the co-payment rate for Xtandi was adjusted from 30% to 5%, canceling out the prescription price reduction. Xtandi's sales recovered, with KRW 6.7 billion in this year's Q1 and KRW 7.4 billion in Q2. Also, in June, Xtandi expanded its indication to non-metastatic hormone-sensitive prostate cancer (nmHSPC), which is expected to increase prescription sales in the future. Sales trend of ARTA treatments used to treat prostate cancer (UBIST report reconstructed by Daily Pharm). With the expanded indications, Xtandi became the only ARTA treatment that can be prescribed to treat all prostate cancer stages following biochemical recurrence, including hormone-sensitive, castration-resistant, non-metastatic, and metastatic. An Astellas official said, "Xtandi is the only prostate cancer treatment that can be used to treat hormone-sensitive HSPC at early stages regardless of metastasis, non-metastasis, and the number of metastases." He said, "When the indication to treat nmHSPC was recently approved, the company focused on marketing to raise awareness of the importance of early ARTA combination therapy to treat HSPC." Erleada chasing Xtandi…Janssen, "To provide patient-individualized treatment option" Although Xtandi is the market leader, Janssen's Erleada, which has been reimbursed since last year, rapidly generates prescription sales. Erleada generated prescription sales of KRW 3.4 billion in last year's Q4. Its sales grew with a bigger margin, with KRW 4.8 billion in this year's Q1 and KRW 7 billion in Q2. Its prescription sales in Q2 were close to Xtandi. Analysis suggests that the close competition may be due to Xtandi's drug price reduction. Erleada appears to be established in clinical practice, as it has been a year since receiving reimbursement. On the other hand, the prescription sales of Janssen's Zytiga (abiraterone) have decreased after the introduction of generic versions. Last year, prescription sales for Xytiga decreased from KRW 4.9 billion in Q3 to KRW 3.5 billion in Q4. In this year's Q1 and Q2, it generated KRW 3.1 billion due to expanded prescriptions of competitor drugs and the introduction of generic products. After Hanmi Pharm's Abiterone was approved last year, generic version of Xytiga were introduced to the market for Xytiga. Furthermore, Hanmi Pharm launched the combination drug Abiterone Duo in February, and Ace Pharmaceuticals obtained approval for Amaron. With more competitors, Xytiga's prescription sales are expected to decrease even more. Changes to the reimbursed price of ARTA treatments used to treat prostate cancer (HIRA report reconstructed by Daily Pharm). Janssen plans to focus on expanding treatment options for prostate cancer sub-types to compete against Xtandi, which has broad indications. Janssen's prostate cancer treatment portfolio consists of Xytiga, Erleada, and Akeega. Akeega was approved in September of last year. Akeega is a treatment for BRCA1/2 mutation-positive metastatic castration-resistant prostate cancer. It is a combination drug combining abiraterone and the PARP inhibitor ingredient niraparib. Prescription sales of individual drugs may be lower than those of Xtandi, but the company's strategy is to face the competition by diversifying treatment options. A Janssen official said, "For the treatment of prostate cancer in South Korea, we have been putting efforts to provide various individualized treatment options and improve treatment accessibility." "We will strive to work as a market leader to provide Korean patients with prostate cancer with optimized treatment by expanding treatment options through Erleada and Xytiga and expanding our portfolio, including Akeega.
- Opinion
- [Reporter's View] Delays in medication switching for eczema
- by Eo, Yun-Ho Aug 13, 2024 05:48am
- When patients switch from their current medication to a different medication, the switched products are not covered by insurance reimbursement. This non-reimbursed status of medication switching has been a long-standing issue in South Korea. The field most impacted is atopic dermatitis. Treatment options had been limited for atopic dermatitis, but various new drugs became available over the past years. New medications, such as interleukin (IL)-inhibitors and JAK inhibitors, have relieved patient burdens, and fortunately, these drugs are listed for reimbursement. However, an issue related to medication switching recently surfaced. When a patient switches prescriptions to a different medication after initially using biologic mediation, such as interleukin (IL)-inhibitors, or oral medication, such as JAK inhibitors, reimbursement is no longer provided. As a result, patients cannot easily switch to a different drug when they experience adverse reactions during treatments or do not benefit from the drugs. Individuals respond differently to all drugs. This could be due to genetic differences or various factors such as age, gender, and race. Having many treatment options means that patients can choose from a pool and anticipate other possibilities when they do not respond to a particular drug. The current Korean policy may be limited to offering this type of 'expectancy.' Additionally, this policy leads to prescription bias. Conventional biologics are more expensive than chemically manufactured medicines. If two medications have equivalent therapeutic status but vary significantly in price, and medication switching is not covered, most patients may be biased towards biologics. In the first half of the year, the Korean Atopic Dermatitis Association stepped forward. The Association submitted an opinion report to the Ministry of Health and Welfare (MOHW) demanding medication switching in treating atopic dermatitis. Furthermore, the Association clarified that there are no differences in therapeutic status between biologic agents and oral medication through the guideline revision made in 9 years. The government initiated a review. Presumably because it has decided that demands and needs are precise. Previously, the government had hesitated to provide reimbursement approval for medication switching due to the "shortage of scientific evidence regarding medication switching." Of course, medication switching is not backed up by clinical studies. However, South Korea is the only country not approving medication switching. Indeed, a study cannot be conducted every time new drugs are introduced and cannot wait for real-world data or other literature review. Ultimately, the speed matters. We have records of wasting close to 10 years in approving reimbursement for medication switching of TNF-alpha inhibitors to treat rheumatoid arthritis. Medication switching of JAK inhibitors in other autoimmune diseases, including ankylosing spondylitis, is already allowed. If the government hesitates, it will take a long time. Furthermore, companies must put efforts into increasing volume of use and finance.
- Company
- Nabota accounts for 24% of Daewoong’s ETC sales
- by Chon, Seung-Hyun Aug 13, 2024 05:47am
- Daewoong Pharmaceutical's botulinum toxin 'Nabota' continued its upward sales trend. The company's quarterly revenue exceeded KRW 50 billion for the first time, led by the company’s growth in overseas markets. Nabota’s share of Daewoong's specialty drug sales reached nearly 25 percent, driving the company's performance. According to Daewoong Pharmaceutical on the 12th, Nabota generated sales of KRW 53.1 billion in Q2, up 62.4% year-on-year. This is the first time since its launch that the drug’s quarterly sales exceeded KRW 50 billion, surpassing the previous record of KRW 42.6 billion set in Q1 last year in less than a year. Nabota’s sales increased 74.7% in 2 years, compared to sales of KRW 30.4 billion in Q1 2022. Daewoong Pharmaceutical Sales of Nabota in the U.S. market continued to rise steadily. Daewoong Pharmaceutical's sales partner for Nabota, Evolus, reported sales of USD 66.9 million (approximately KRW 92 billion) in Q2, up 35.6% from the USD 49.35 million made year-on-year. This marks the fifth consecutive quarter Evolus renewed its revenue record since the company reported USD 49 million in Q2 last year. Nabota's export performance has begun to surge since the closure of its strain theft lawsuit with Medytox, which began in 2019, and the established trust in Nabota based on the accumulated U.S. experience. In February 2021, Medytox entered into a settlement agreement with Daewoong Pharmaceutical's U.S. partners Evolus and AbbVie for the sale of Nabota (U.S. trade name Jeuveau) in the United States. Under the agreement, Medytox and AbbVie received the rights to continue marketing and distributing Jeuveau in the U.S. for certain payments from Evolus. Evolus is also looking to expand into the European market, having launched Nabota in Spain in June. In Europe, Nabota is currently available under the brand name Nuceiva and is now available in the U.K., Germany, Austria, Italy, and the U.S., in addition to Spain. In June, the company received marketing authorization for Nabota from Argentina's National Administration of Drugs, Food and Medical Devices (ANMAT), accelerating its international expansion as well. Nabota will be launched in Argentina under the brand name Clodew in Q4 this year through Daewoong’s local partner Oxapharma. Daewoong Pharmaceutical currently has botulinum toxin products licensed in more than 70 countries worldwide and partnerships in more than 80 countries. As a result, Nabota's presence in Daewoong's sales is also gradually expanding. In Q2 last year, Nabota’s sales accounted for 24.4% of Daewoong's KRW 218 billion in specialty drug sales, the largest share ever. Among the products developed and sold by Daewoong Pharmaceutical, Nabota has generated the largest sales volume. Daewoong's sales of specialty drugs increased 11.7% in 3 years from KRW 181 billion in Q2 2021, while Nabota’s sales more than tripled in the same period. Nabota's share of the company's specialty drug sales has nearly tripled in 3 years from 8.5% in Q2 2021. Nabota’s share in Daewoong Pharmaceutical's specialty drug sales exceeded 10% in Q3 2020 for the first time. After surpassing 20% in Q1 last year at 20.6%, it remained in the 10% range until Q1 this year. However, with the surge in Nabota’s sales, the drug accounted for more than a quarter of the company’s specialty drug sales in Q2. More recently, the company’s new drug Fexclue has also contributed significantly to company sales. In Q2, sales of Fexuclue more than doubled year-on-year to KRW 33.2 billion. Fexclue is a potassium-competitive acid blocker (P-CAB) drug that treats GERD. Fexuclue was approved by the MFDS in December 2021 and began full-scale sales in July 2022 after being listed for reimbursement on Korea’s health insurance benefit list. Nabota and Fexuclue jointly generated KRW 86.3 billion in sales in Q2, accounting for 39.6% of the company's specialty drug sales. Daewoong Pharmaceutical set a new performance record thanks to the steep growth of its self-developed drugs. On a standalone basis, Daewoong Pharmaceutical's Q2 revenue increased 6.0% year-on-year to KRW 325.5 billion, and operating profit increased 37.1% to KRW 49.6 billion. Both the revenue and operating profit in Q2 were the largest in the company's history. The operating profit margin as a percentage of sales was 15.2%, a significant improvement from the 11.8% it had made in the same period of the previous year, showing high performance. Daewoong Pharmaceutical's operating profit on a consolidated basis was KRW 42.3 billion in Q2, up 5.6% year-on-year, the largest ever. Revenue increased 3.0% year-on-year to KRW 360.5 billion, the second-highest amount ever recorded after Q4 of last year.
- Company
- 'High immunity vs. high dose’ flu vaccine for older adults
- by Hwang, Byung-woo Aug 13, 2024 05:47am
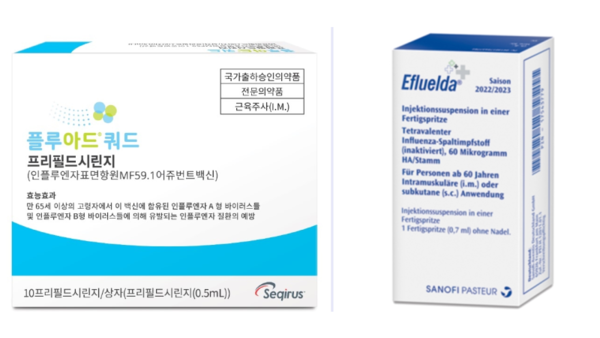
- Equipped with their respective flu vaccines specialized for people aged 65 and older, Sanofi and CSL Seqirus are seeking to expand their market share in Korea’s influenza (flu) vaccine market, which is driven by vaccines registered in the National Immunization Program (NIP). CSL Seqirus was the first to enter this market last year, followed by Sanofi, which newly entered the market this year. The companies will face off for the first time in the 2024-25 flu season. Each is expected to target, highlighting their respective characteristics, high immunogenicity and high dose. (From the left) Pic of Fluad Quadrivalent, Efluelda According to industry sources on the 13th, ‘Efluelda,’ Sanofi’s high-dose influenza vaccine for people aged 65 and older, will be launched in time for this year’s flu season. Efluelda contains 4 times more the amount of antigens compared with conventional vaccines and has a mechanism of action that elicits a stronger immune response, preventing 24.2% more infection compared to the standard-dose vaccine. Previously, CSL Seqirus’s Fluad Quad was the first flu vaccine to be launched for people aged 65 and older. It was approved by the Ministry of Food and Drug Safety in 2022 and launched in time for the 23/24 season last year. Fluad Quad is a quadrivalent influenza vaccine that includes the immune booster 'MF59' and was developed by adding one type of influenza B virus to the company’s existing trivalent influenza vaccine Fluad. It offers improved size and breadth of immune response through the immune booster in older adults. The subsequent entry of Efluelda into the market, which was preemptively occupied by Fluad Quad, has ignited competition in the 65+ age group. While the inclusion of the influenza vaccine in the NIP could be a hurdle, the two companies believe there is an unmet need for flu protection amongst the elderly in Korea. In fact, despite the high rates of influenza immunization among people aged 65 and older, two-thirds of all influenza deaths in Korea occur in people 60 and older. In particular, older adults are known to be particularly vulnerable to influenza infection and complications due to decreased immune function and comorbidities. They also receive less protection after vaccination than younger adults. "While the majority of influenza vaccinations for people aged 65 and older are administered through NIP, the company believes there is still significant market development potential given the lower vaccine efficacy and risk of complications in older adults," said a Sanofi representative. "The need for high immunogenic influenza vaccination option has been increasing amongst older adults in Korea, with the Korean Society of Infectious Diseases' revision to the Adult Immunization Guideline last year recommending high immunogenic influenza vaccination for those aged 65 and older," said a CSL Seqirus representative. Fluad Quad was approved in 2022 and launched on the market last year Flu vaccination for older adults enters NIP...companies ponder over private market marketing challenges NIP However, due to the high share of NIP vaccinations amongst total influenza vaccinations, the private, non-reimbursed market for those aged 65 and older is limited. As a result, both Sanofi and CSL Seqirus would need to highlight the value of their vaccines, while holding in check the competition between products. In its second season on the market, Fluad Quad is expected to be available in the country from mid-September, at a similar volume to last year. CSL Seqirus’s marketing keyword for this season is 'filial piety'. The company is focusing on communicating the differentiated superiority of Fluad Quad compared to the existing egg-based vaccine and is planning activities to express the feelings of sons and daughters who care about their parents' health. In particular, before the launch of the influenza vaccine, the company emphasized its strengths over its competitor, announcing the results of a study on the relative vaccine effectiveness (rVE) of trivalent immune-boosting influenza and trivalent high-dose influenza vaccines. "While high-dose influenza vaccines simply increase the amount of antigen, Fluad, an immune-boosting influenza vaccine, is different as it improves the immune response through MF59, an adjuvant developed through our company’s proprietary technology, to enhance the prevention effect," emphasized a CSL Seqirus representative. While Sanofi did not disclose specific volumes, the company said it is preparing for full-scale marketing activities, including national lot release, for Efluelda. The marketing keyword for Efluelda is "flu prevention, more than just protection" and will focus on its ability to address the unmet needs of older adults in terms of flu infections and hospitalizations from complications. "We are taking a deliberate and systematic approach to address the unmet need for flu prevention in the elderly," said a Sanofi representative. "We are planning a range of promotional activities targeting healthcare providers as well as consumers, as the high-dose vaccine has been shown to provide greater protection and reduced hospitalization rates compared with conventional vaccines."

- Company
- Hanmi’s Rolvedon posts sales of KRW 20.6 billion in Q2
- by Son, Hyung-Min Aug 13, 2024 05:47am
- Sales of Rolvedon (Korean brand name: Rolontis), a new anti-cancer drug developed by Hanmi Pharmaceutical, are showing signs of recovery in the U.S. market. Hanmi Pharmaceutical's U.S. partner Assertio plans to increase Rolvedon’s market share through new clinical trials that could ensure the drug’s advantage in convenience of administration. According to Assertio on the 12th, Rolvedon generated USD 15.1 million in U.S. market sales in Q2 this year, down 28.1% YoY, but up 4.1% from the previous quarter. Rolvedon is a neutropenia treatment developed by Hanmi Pharmaceutical and was approved as the 33rd homegrown new drug in Korea in March 2021. Hanmi Pharmaceutical and its U.S. partner Spectrum (now Assertio) obtained U.S. Food and Drug Administration (FDA) approval for Rolvedon in September 2021. Hanmi Pharmaceutical transferred Rolvedon’s technology to the U.S. company Sepctrum in 2012. Assertio acquired Spectrum and Rolvedon in April last year. Assertio specializes in the development of inflammation treatments, including the non-steroidal anti-inflammatory drug indocin and the orally disintegrating film-based drug Sympazan. The company sought to enter the anticancer market with Rolvedon. The drug is administered to prevent or treat neutropenia in cancer patients who receive myelosuppressive chemotherapy. As a granulocyte colony-stimulating factor (G-CSF) class drug, it stimulates the granulocyte to increase neutrophil production, similar to ‘Neulasta (pegfilgrastim).’ Changes in quarterly sales of Rolvedon (Unit: USD 1 million) Since its launch in October 2022, the drug generated USD 10.1 million in Q4 sales. In December of the same year, Rolvedon was included in the National Comprehensive Cancer Network (NCCN) febrile neutropenia prevention and treatment guidelines. Since then, Rolvedon’s sales continued to grow, generating USD 15.6 million in Q1 and USD 21 million in Q2 last year. Rolvedon’s sales growth slowed in Q3 last year, posting sales of USD 8 million, a 62.0% decrease from the previous quarter. The decline was primarily due to new reimbursement terms applied to Rolvedon. The new reimbursement system, which has been in place since April last year, is reportedly less favorable than the terms that were applied upon its launch. Rolvedon’s sales rebounded in Q4 last year, generating USD 10 million in revenue, and its sales continued to grow in Q1 and Q2 of this year, posting USD 14.5 million and USD 15.1 million, respectively. Assertio attributed the rebound to new accounts won with its new commercialization strategy. Since its launch in the U.S. in Q4 2022, Rolvedon has generated USD 95.3 million (KRW 130 billion) in cumulative sales. Company completes patient enrollment for a same-day dosing clinical trial for Rolvedon...to compete with Neulasta Assertio made a bid to Rolvedon’s competitor Neulasta with a same-day dosing trial. One of the disadvantages of using neutropenia treatments like Neulasta is that the drug can't be administered until 24 hours after chemotherapy, which increases the number of hospital days for patients. Amgen and Kyowa Kirin, the developers of Neulasta, are defending the market with the launch of Neulasta Onpro, which is designed to be worn on the day of chemotherapy and automatically deliver Neulasta the next day. Neulasta OnPro is designed to be attached to a patient's body the same day they receive chemotherapy, and then automatically deliver the drug the next day. Assertio plans to compete with Neulasta Onpro with a same-day dosing trial of Rolvedon. Assertio is currently enrolling patients in its Phase I same-day dosing trial for Rolvedon. Assertio aims to present initial same-day clinical data at an international academic conference within the year.
- Company
- JAK inhibitors market size rose 54%↑over the year
- by Kim, Jin-Gu Aug 12, 2024 05:55am
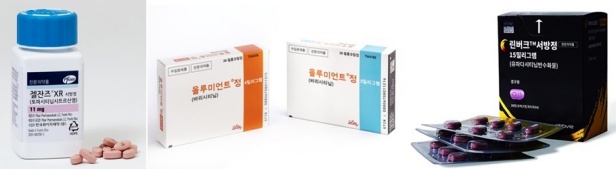
- (Clockwise from upper left) product photos of Jyseleca, Cibinqo, Rinvoq, Olumiant, and Xeljanz. The market for Janus Kinase (JAK) inhibitors, oral medicines used to treat autoimmune diseases, is growing rapidly following the approval of major medicines for expanded indications. In the first half of the year, the market for JAK inhibitors reached KRW 27.5 billion in outpatient sales, up 54% year over year (YoY). Abbvie's 'Rinvoq (upadacitinib)' occupies the No.1 slot among the major products with a considerable margin over other products. Lily's 'Olumiant (baricitinib),' Pfizer's 'Xeljanz (tofacitinib)'·'Cibinqo (abrocitinib),' and Eisai's 'Jyseleca (filgotinib)' are increasing prescription performance. The JAK inhibitors market grew 54% over the year…market size expected to be worth KRW 50 billion by the end of 2024 According to the pharmaceutical market research firm UBIST on August 9th, the outpatient market size for JAK inhibitors in the first half of 2024 is worth KRW 27.5 billion, up 54% YoY compared to KRW 17.8 billion last year. JAK inhibitors are medicines used to treat autoimmune diseases, including rheumatoid arthritis and atopic dermatitis. They work inhibiting inflammatory cytokines, which in turn inhibit inflammation, pain, and cell activation. After the launch of Xeljanz in 2015, Olumiant and Rinvoq were introduced to the race in 2019 and 2021, respectively. Cibinqo and Jyseleca followed last year. South Korea JAK inhibitors market outpatient prescription sales (unit: KRW 100 million, source: UBIST). Eisai’s Jyseleca (red), Pfizer’s Cibinqo (purple), Abbvie’s Rinvoq (light blue), Lily’s Olumiant (blue), and Pfizer’s Xeljanz (dark blue). After new drugs launched one after another, the market is expanding rapidly. JAK inhibitors market worth KRW 12.5 billion in 2019 has expanded to KRW 18.7 billion in 2020, KRW 25.5 billion in 2021, KRW 33.5 billion in 2022, and KRW 40 billion last year. The market recorded KRW 27.5 billion just for the first half of this year and is expected to top KRW 50 billion by the end of 2024. The prescription sales of the market leader Rinvoq doubled over the year…a substantial margin compared to No.2 and No.3 Among major products, Rinvoq is at the top of the ranking. The prescription sales of Rinvoq for the first half of the year amounted to KRW 11.2 billion, a 2.2-fold increase from KRW 5.2 billion YoY. Following Rinvoq, Olumiant ranked No.2 with KRW 8 billion in the first half of the year. It has seen a 28% increase over the year compared to KRW 6.2 billion YoY. During the same period, the prescription sales of Xeljanz increased 8%, from KRW 6.4 billion to KRW 6.9 billion. These three products competed against each other for the No.1 slot. Xeljanz, the first drug launched, was a market leader until the first quarter of last year. However, Olumiant surpassed Xeljanz and occupied the No.1 slot in the second and third quarter of last year, with a marginal difference. Quarterly prescription sales of JAK inhibitors (unit: KRW 100 million, source: UBIST). Xeljanz (dark blue), Olumiant (blue), and Rinvoq (light blue). From the fourth quarter of last year, Rinvoq's sales grew rapidly. Rinvoq's prescription rose substantially in the fourth quarter of last year, and Rinvoq, which used to rank No.3, became No.1. Furthermore, its prescription sales increased by a greater margin in first and second quarters of this year. Rinvoq has surpassed the prescription sales of Olumiant and Xeljanz, dominating the market. Rinvoq experienced rapid growth in sales following approval for "adolescent's atopic dermatitis' indication…in progress for adding 'children's atopic dermatitis' Analysis suggests that expanded indication have contributed to Rinvoq's rapid growth, especially after it was approved for the indication to treat adults and adolescents 12 years of age and older with atopic dermatitis. Rinvoq is indicated for the treatment of ▲rheumatoid arthritis, ▲psoriatic arthritis, ▲ankylosing spondylitis, ▲atopic arthritis, ▲ulcerative colitis, and ▲Crohn's disease. Olumiant is indicated for the treatment of ▲rheumatoid arthritis, ▲atopic dermatitis, and ▲alopecia areata. Xeljanz is indicated for the treatment of ▲rheumatoid arthritis, ▲psoriatic arthritis, and ▲ankylosing spondylitis. Rinvoq and Olumiant's increased prescription sales were driven by atopic dermatitis. However, these two differ in the range of usage. Rinvoq can be used to treat adults and adolescents of 12 years and older with moderate to severe atopic dermatitis, while Olumiant can be used to treat adults with moderate to severe atopic dermatitis. Additionally, a Phase 3 clinical trial for Rinvoq is in progress, targeting children of 2 to 12 years with atopic dermatitis. When the clinical trial concludes and receives approval for an expanded indication, Rinvoq's prescription sales will rise even higher. Cibinqo and Jyseleca, launched last year, are growing in market impact. Pfizer launched Cibinqo with reimbursement coverage in July of last year, as a Xeljanz follow-up. Cibiqo recorded KRW 700 million in prescription sales in last year's second half and generated KRW 1.2 billion in this year's first half. Cibinqo received approval for the indication to treat atopic dermatitis, the market dominated by Rinvoq and Olumiant. It is expected to have rapid growth, considering that it secured an indication to treat moderate to severe atopic dermatitis in adults and adolescents aged 12 years or older, similar to Rinvoq. Jyseleca was launched in November of last year as the fifth JAK inhibitor. It is indicated for the treatment of rheumatoid arthritis and ulcerative colitis. For the first half of 2024, the prescription sales for Jyseleca were KRW 300 million.
- Company
- Columvi can be prescribed at general hospitals in KOR
- by Eo, Yun-Ho Aug 12, 2024 05:55am
- ‘Columvi,' the first bispecific antibody treatment option for lymphoma, may be prescribed at general hospitals in Korea. According to industry sources on the 9th, Roche Korea's CD20-CD3 bispecific antibody for diffuse large B-cell lymphoma (DLBCL) Columvi (glofitamab) passed Samsung Medical Center’s drug committee review. However, Columvi is currently a non-reimbursed drug. Its reimbursement application was reviewed by the Cancer Disease Deliberation Committee in July but was unable to set reimbursement standards at the time. Whether the company will quickly reorganize and reapply for coverage is gaining interest. Columvi was approved in Korea last December for the treatment of adult patients with relapsed or refractory diffuse large B cell lymphoma (DLBCL), after two or more lines of systemic therapy. The drug is a third-line treatment option for DLBCL, like Novartis’s chimeric antigen receptor (CAR)-T-cell therapy Kymriah (tisagenlecleucel). The two drugs have different benefits; therefore the choice will likely be based on each patient's condition and circumstance. Columvi demonstrated efficacy in the Phase I/II NP30179 trial in 155 patients with relapsed or refractory DLBCL after two or more prior systemic therapies. Results showed that Columvi achieved a complete response (CR) of 40% and an overall response rate(ORR) of 52%. The efficacy was also consistent across all subgroups. The most common adverse event was cytokine release syndrome (CRS). Adding to the encouraging data, at the 2024 Congress of the European Hematology Association (EHA 2024), the company unveiled the results of the Phase III STARGLO study, which demonstrated an improvement in overall survival (OS) with Columvi. The STARGLO study enrolled patients with relapsed or refractory (R/R) diffuse DLBCL who were not eligible to receive an autologous stem cell transplant after one or more prior systemic therapies, or who had received two or more prior systemic therapies. In the primary analysis (median follow-up 11.3 months), Columvi and gemcitabine+oxaliplatin (GemOx) combination significantly improved the primary endpoint of OS with a 41% lower risk of death compared to rituximab+GemOx. Seok Jin Kim, Professor of Hematology and Oncology at Samsung Medical Center, said, "There had been much unmet need in DLBCL for more effective third-line treatment options for patients who fail first-line or experience repeated relapses. We expect the introduction of Columvi to significantly improve the outcomes for patients with relapsed or refractory lymphoma in Korea."
- Company
- Anticancer drug 'Alecensa' expected to win nod
- by Eo, Yun-Ho Aug 12, 2024 05:55am
- Anticancer drug The ALK anticancer drug Alecensa is anticipated to receive approval for an additional indication as an adjuvant therapy for treating lung cancer. Sources said that the review for indication approval by the Ministry of Food and Drug Safety (MFDS) is in its final stage for Alecensa (alectinib), Roche Korea's anaplastic Lymphoma Kinase (ALK)- targeted anticancer agent, as a post-operative adjuvant therapy for patients with early-stage non-small cell lung cancer (NSCLC). The indication of Alecensa has been approved worldwide, including by the U.S. FDA in April and the European Commission (EC). The basis of indication for the adjuvant therapy for early-stage NSCSC was the Phase 3 ALINA study, which demonstrated the efficacy. The study compared Alecensa to platinum-based chemotherapy to confirm the potential use of the drug as post-operative chemotherapy for treating patients with early-stage NSCLC. The trial enrolled 257 patients with stage IB to IIIA ALK-positive NSCLC who are 18 years old and older. The patients were randomly assigned to the Alecensa 600 mg treatment group (twice daily) and the platinum-based chemotherapy treatment group (max. 4 times every 3 weeks). The primary endpoint of the study was set as disease-free survival (DFS), and the secondary endpoints were overall survival (OS), the Central Nervous System-DFS (CNS-DFS), and the safety. The clinical results showed that the Alecensa treatment group had a 93.8% DFS at 2 years, which was higher than the 63% DFS of the platinum-chemotherapy treatment group. The 2-and 3-year CNS-DFS for the Alecensa treatment group were 98.4% and 95.5%, respectively. These measures were higher than the respective rates of 85.8% and 79.7% for the platinum-based chemotherapy treatment group. Furthermore, the Alecensa treatment group had a reduced risk of CNS disease progression or death by 78%. Meanwhile, adjuvant therapy with 'Tagrisso (osimertinib)' is commonly used to treat EGFR-positive NSCLC based on the ADAURA study, which demonstrated a superior effect over the platinum-based chemotherapy. Since Alecensa has shown to be more effective than the platinum-based chemotherapy, its use in clinical practice is expected to be widespread.

- Policy
- Reimb for Paxlovid still pending amid COVID-19 resurgence
- by Lee, Tak-Sun Aug 12, 2024 05:55am
- Product photo of Paxlovid. As the COVID-19 treatment supply becomes an ongoing issue amid a virus resurgence, clinical practices are frustrated by delayed reimbursement coverage of 'Paxlovid (nirmatrelvir‧ritonavir, Pfizer Korea).' The drug reimbursement application for Paxlovid was submitted to the Health Insurance Review and Assessment Service (HIRA) in early October of last year. After that, HIRA has yet to finish the evaluation. On August 8th, Paxlovid was not considered for HIRA's Drug Reimbursement Evaluation Committee (DREC) review. Initially, Pfizer aimed to acquire reimbursement for Paxlovid for the first half of the year, but it has yet to be considered for the DREC review 10 months after submission. During this period, Pfizer submitted response letters to HIRA six times. The company recently requested HIRA an extended response letter due date. While the reimbursement for Paxlovid is being delayed, the Korean Disease Control and Prevention Agency (KDCPA) discontinued the free-of-charge program in May. Now, patients are required to pay KRW 50,000, which is approximately 5% of the drug price, for the drug. However, it is still provided free of charge for medical reimbursement benefit recipients and those eligible for co-payment reduction due to having the second-lowest income. When the drug became available for co-payment, some clinical practices prescribed Paxlovid to adult patients under different conditions. The basis for Paxlovid approval in South Korea is its efficacy and effectiveness in treating adults with mild to moderate symptoms who are at a high risk of developing severe COVID-19, including hospitalization and death. As a result, prescribing it to adults with mild symptoms still meets the efficacy and effectiveness criteria. However, when the government provided Paxlovid free of charge, it was only used for patients who were senior-aged and had severe symptoms. It seems that when the drug became available for co-payment, clinical practices mistakenly thought it had reimbursement criteria and prescribed it as approved. Recently, the Korea Disease Control and Prevention Agency (KDCPA) officially requested that clinical practices prescribe oral COVID-19 medicines, including Paxlovid, only to high-risk patients who show symptoms. Those who can be prescribed are patients over 60 years old and 12 years old (Paxlovid) or 18 years old and older (Lagevrio) who are immune compromised or with underlying diseases. Additionally, it must prescribed within 5 days of showing symptoms and for patients not needing oxygen therapy. The KDCPA said, "There are no changes to who can be prescribed COVID-19 medicines before or after issuing co-payment in May." They explained, "It can only be prescribed in clinical practices designated for COVID-19 medicines prescription." To prevent such confusion, some have pointed out the necessity of prescription criteria through reimbursement coverage, as well as agreements with pharmaceutical companies for the efficient supply of drugs. A HIRA official said, "Paxlovid will be internally evaluated soon and considered for the DREC review."
- Opinion
- [Column] On patent strategies post Amgen v. Sanofi dispute
- by Kim, Jin-Gu Aug 12, 2024 05:55am
- One of the most significant events that occurred in the bio-pharmaceutical sector in recent years was the ruling made on the Amgen vs. Sanofi patent dispute in the United States. In May last year, the U.S. Supreme Court issued its final ruling on the patent dispute between the two biotech giants. The case received special attention, as the dispute over the validity or invalidity of patents lasted for nearly a decade in the KRW 1 trillion drug market and the first in a long time the U.S. Supreme Court ruled on the 'enablement requirement' of a patent. In 2011, Amgen and Sanofi each patented an antibody for the treatment of hyperlipidemia that binds to the PCSK9 protein, and developed Repatha and Praluent, respectively. Three years later, in 2014, Amgen registered two additional patents for antibodies that bind to the PCSK9 protein and immediately filed a patent infringement lawsuit against Sanofi. In response, Sanofi sought to invalidate both of Amgen's patents, and the dispute came to a head. Amgen was able to file its patent infringement lawsuit in 2014 instead of 2011 for a reason. Amgen's 2011 patent limited antibodies to an amino acid sequence, whereas the 2014 patent claimed antibodies more broadly. Amgen’s patent consists of an antibody that ▲binds to a specific amino acid or epitope of PCSK9; and ▲ prevents PCSK9 from binding to the LDL receptor. Claiming an entire group of antibodies that exhibit this specific function is called a “genus type” claim, which can include millions of individual species antibodies. As a result, genus-type claims have a very broad scope. Despite making such a broad claim, Amgen’s patent specification only disclosed: i) 26 amino acid sequences for antibodies that would block or inhibit PCSK9, and ii) two alternative methods (the roadmap, conservative substitution) for arriving at the remainder of the genus. The court found that the 26 exemplified antibodies were not sufficient to readily produce the entire genus of antibodies and that Amgen's 2 methods constituted separate research projects. The experts ruled Amgen's patents invalid and that they did not meet the enablement requirement because it would require extensive experimentation and trial and error to reproduce the invention. The Supreme Court’s ruling does not overturn the existing view of the enablement requirement, but it does set a clear precedent for the strict application of the requirement, which will have a significant impact on the industry going forward. The Amgen case has several implications. First, it highlights the need for new patent portfolio management. The U.S. Supreme Court's decision has a significant impact on the global patent industry in general. In the biotech sector, especially for antibodies, it will become more difficult to patent an entire genus of antibodies by function. The number of patent applications will naturally increase, and divisional patent application is expected to increase. Furthermore, the treatment of a double patent varies by country, requiring closer attention. A company that discovers a new target should attempt various different types of patent claims. In the bio field, discovering a new target is like finding a gold mine. Therefore, the company should protect the mine by pursuing the broadest claims possible. "Functional claims," like in Amgen’s case, have the advantage of capturing the best features of the target, but they are more likely to be rejected or invalidated. Therefore, rather than pursuing purely functional claims, claims should be drafted in a way that blends functional and structural elements. It's also a good idea to experiment with multi-step claim construction, such as going from broad to narrow. Deciding when to file is a key point in the bio patent application strategy. Historically, patent strategies had been focused on patent maps during the R&D phase, freedom to operate (FTO), or litigation strategies before the product launch, but companies should now pay more attention to the patent application strategy. In the bio sector, it takes a long time to generate experimental data, so it is especially important to decide when to file the patent, after securing how much data. More data can lead to a broader scope of rights but at the cost of a later patent application date. The patent application date should take into account whether the patent is for a platform technology or a follow-on technology, whether it is in the early research or clinical stage, how large the market is for the product, and how much data can be produced. It is recommended that universities, research institutes, and companies plan their patent application strategies according to their data production capacity. The stricter the patent system interprets the enablement requirement, the more favorable it is for global companies. For example, if a global company and a university discover a disease target "at the same time," the global company will have a better chance of obtaining a broad patent because it can produce a lot of data in a relatively short period of time. If a university, research institute, or startup makes an important early stage discovery, one way to reduce the risk is to find a collaborative research partner or license out the technology to increase data production capacity.