- LOGIN
- MemberShip
- 2026-06-20 21:42:03

- Company
- Opdivo·Yervoy combination therapy for HCC
- by Whang, byung-woo Jul 23, 2025 06:08am
- Opdivo, celebrating its 10th anniversary of approval in South Korea, is expected to expand its influence by broadening the indication of its combination therapy with Yervoy to include first-line treatment for hepatocellular carcinoma (HCC). Experts assess that this will become a primary treatment option, given the unmet need in the existing first-line HCC treatment landscape, where tyrosine kinase inhibitors (TKIs) have been the standard of care for over 10 years. Although reimbursement remains a challenge, there is a view that it will become a new standard of care. Ono Pharma Korea and BMS Korea held a press conference on the 22nd of this month, highlighting the expanded indication of Opdivo (nivolumab) and Yervoy (ipilimumab) combination therapy for first-line HCC treatment. Opdivo is a PD-1 (programmed cell death-1) immune checkpoint inhibitor that reactivates the body's immune system to mount an anti-tumor immune response by inhibiting the PD-1 and PD-1 ligand pathways. Dr. Do Young Kim of the Department of Gastroenterology at Severance Hospital First introduced in Korea in 2015 as a second-line treatment for malignant melanoma, as of July 2025, it has secured 24 indications across a total of 11 cancer types. On July 10, the combination therapy of Opdivo and Yervoy received additional approval from the Ministry of Food and Drug Safety (MFDS) for the first-line treatment of unresectable or metastatic HCC. The CheckMate-9DW study, which served as the basis for the first-line approval of the Opdivo-Yervoy combination therapy for HCC, is receiving a positive evaluation for setting lenvatinib as a comparator drug. Dr. Do Young Kim of the Department of Gastroenterology at Severance Hospital explained, "Most treatments approved for first-line HCC treatment have used sorafenib as a comparator, but this study also set lenvatinib, which is a relatively newer treatment option, as a comparator." He added, "The significance lies in confirming the superiority of the Opdivo+Yervoy combination therapy under conditions more similar to the current clinical environment, as lenvatinib was administered to 85% of the overall comparator arm." The Opdivo+Yervoy combination therapy, based on the CheckMate-9DW study, demonstrated a median overall survival (OS) of 23.7 months, with a median follow-up period of 35.2 months. The comparator group had an OS of 20.5 months, showing that the Opdivo+Yervoy combination arm had a 21% lower risk of death than the comparator arm. The OS rates for the Opdivo+Yervoy combination therapy group at 2 and 3 years were 49% and 38%, respectively, surpassing the comparator group's 39% and 24%. Dr. Kim said, "The CheckMate-9DW study results showed that the improved overall survival benefit of the Opdivo+Yervoy combination therapy was maintained up to the 3-year mark compared to sorafenib and lenvatinib," and added, "This confirms its sustained efficacy as a dual immunotherapy." Dr. Kim also mentioned, "The Opdivo+Yervoy combination therapy showed approximately a threefold improvement in both response rate and complete response rate compared to the comparator group, and the median duration of response was 30.4 months. In other words, patients who respond to Opdiv+Yervoy combination therapy, long-term therapeutic effects of over 2.5 years can be expected." "Opdivo+Yervoy expected to address unmet needs in HCC" Dr. Chang-hoon Yoo, Professor of the Department of Medical Oncology at Asan Medical Center in Seoul, highlighted the possibility that the Opdivo+Yervoy combination therapy could become a new standard of care in HCC, where existing treatments have faced limitations. According to Dr. Yoo, liver cancer (disease code C22) had the second-highest mortality rate among major cancer types in Korea as of 2023, at 11.9%, following lung cancer (21.9%). In the same year, total deaths due to liver cancer amounted to 10,136. Dr. Chang-hoon Yoo, Professor of the Department of Medical Oncology at Asan Medical Center in SeoulDr. Yoo explained, "The 5-year survival rate for advanced HCC is reported to be less than 5%, and TKIs, which have been used as standard first-line treatment for over 10 years in the existing treatment environment, only showed a survival period of around one year." He added, "While immunotherapy-based combination therapies were introduced as new treatments for HCC in 2022, changing the treatment landscape, there was still a demand for additional alternatives in terms of survival duration, response rates, and side effects like variceal bleeding." Currently, Dr. Yoo's opinion is that the approval of Opdivo+Yervoy combination therapy for the HCC indication makes it the first treatment to consider for prescription. Dr. Yoo stated, "The CheckMate-9DW clinical trial results, which showed high response rates and the potential for long-term survival of approximately two years, demonstrated its potential to serve as a new standard of care in first-line HCC treatment in Korea." He added, "The treatment response of Opdivo+Yervoy combination therapy was consistent regardless of the patient's liver function, providing a basis for selecting it as a first-line option considering the patient's condition." He also assessed, "It can be primarily considered for patients whose main treatment goal is long-term survival." Notably, Dr. Yoo anticipated no concerns regarding safety, given that the Opdivo+Yervoy combination therapy has already accumulated sufficient clinical experience through the prior application system for several years. Dr. Yoo said, "The safety profile was similar to the existing profiles of each drug, and adverse events were mostly manageable," and added, "Opdivo and Yervoy have accumulated sufficient clinical experience in combination therapy for HCC through the prior application system for several years now." Dr. Yoo further added, "Immune-related adverse events can be safely managed through early detection and monitoring, and in actual clinical studies, cases where immune-related adverse events led to treatment discontinuation were rare. Concerns regarding adverse event management are expected to be minimal."
- Policy
- Supply shortage of Parkinson’s drug Equfina is expected
- by Lee, Hye-Kyung Jul 23, 2025 06:08am
- Eisai Korea's Parkinson's disease treatment ‘Equfina Film-Coated Tab 50mg (safinamide mesilate)’ is expected to be in short supply for the next month, causing inconvenience to patients. In particular, although there are drugs with similar efficacy to Equfina, there are no direct therapeutic substitutes available with the same ingredients. The company reported the supply shortage to the Ministry of Food and Drug Safety on the 21st and announced today (from the 22nd) that a temporary supply shortage of Equfina is expected until August 31. The reason for the shortage is an issue with the release testing. Eisai stated, “Due to issues with the external testing agency, release testing has not been conducted, and a temporary supply disruption is expected once existing inventory is depleted.” Eisai explained, “While there are alternative medications with similar efficacy used for Parkinson's disease treatment, Equfina is a new drug, so there are currently no direct therapeutic substitutes. We will work closely with the testing agency to resolve the supply shortage as soon as possible.” Currently, there are no generic versions of Equfina available in the domestic market. However, companies such as Myung In Pharm, Bukwang Pharmaceutical, and Samil Pharmaceutical filed patent challenges. Myung In Pharm received approval on the 17th to initiate a bioequivalence test for its Equfina generic and is now proceeding with product development. Equfina was approved by the Ministry of Food and Drug Safety on June 24, 2020, as a once-daily levodopa adjunctive therapy for Parkinson's disease patients. Equfina is a new third-generation MAO-B (monoamine oxidase-B) inhibitor that acts on both dopaminergic and non-dopaminergic signaling pathways. In a Phase III clinical trial, it demonstrated significant improvements in both motor and non-motor symptoms in Parkinson's disease patients experiencing motor fluctuations. The current standard of care for Parkinson’s disease is levodopa, but it has been reported that approximately 75% of patients may develop complications after long-term (5 years or longer) use of levodopa. Equfina is used as an adjunctive therapy to levodopa, increasing the duration of action of levodopa, which may decrease with long-term use. Equipina is initiated at a dose of 50 mg once daily, with the option to increase the dose to 100 mg once daily (two 50 mg tablets) based on individual patient response and tolerability. The incidence of adverse reactions following Equipina administration was similar to that of placebo, with most (over 90%) being mild to moderate in severity. Even after two years of long-term administration, it demonstrated a safety profile similar to those observed in previous studies.

- Company
- The driving force behind Lundbeck's long-term innovation
- by Cha, Jihyun Jul 23, 2025 06:07am
- "Lundbeck is a publicly listed company, but 70% of its shares are held by the Lundbeck Foundation. This structure has provided a foundation for Lundbeck to focus on its core value without being swayed by short-term stock price fluctuations. The Lundbeck Foundation reinvests the dividend income it receives from the operating company (Lundbeck) in research and development in the fields of brain disorders and neuroscience." Lan Ding, Vice President of Corporate and Portfolio Strategy, spoke about the company’s governance system at Lundbeck's headquarters in Valby, Copenhagen, Denmark. Ding explained that the stable investment structure backed by the foundation acts as a catalyst for accelerating innovation through a virtuous cycle. Lan Ding, Vice President of Lundbeck Corporate and Portfolio Strategy Lundbeck is a pharmaceutical company that has focused exclusively on brain disease treatments since its establishment in 1915. It has built a pipeline covering the entire spectrum of central nervous system disorders, from mental illnesses such as schizophrenia and depression to neurological disorders such as migraine and epilepsy. As of the closing price on the 10th, the company's market capitalization stood at DKK 32.9 billion (approximately KRW 8 trillion), ranking among the top 20 companies on the Danish stock exchange. Ding holds a master's degree in supply chain management from Lancaster University in the UK and an EMBA from London Business School (LBS). She has worked in strategy and portfolio management at global pharmaceutical companies such as GlaxoSmithKline (GSK). She joined Lundbeck in January this year as vice president of corporate and portfolio strategy, and has been directing the company's mid- to long-term growth strategy. What has enabled Lundbeck to continue innovating for over 100 years? Ding cited four factors: ▲a focus on neuroscience, ▲continuous R&D investment, ▲open innovation, and ▲a foundation-owned governance structure. First, Ding emphasized the company's strategy of dedication and focus on CNS. “Our deep and unwavering focus on neuroscience is the foundation of Lundbeck's innovation,” said Ding. “Reinvesting more than 20% of our annual revenue in R&D based on scientific data and unmet medical needs is also a key factor.” Open innovation is another pillar supporting Lundbeck's innovation. Lundbeck is expanding its scientific scope and accelerating innovation by forming strategic partnerships with various stakeholders, including academia, biotech companies, and patient groups. Lundbeck is also actively pursuing mergers and acquisitions (M&A) to accelerate growth, not just technology adoption. A notable example is last year's acquisition of Longboard Pharmaceuticals for USD 2.6 billion, when the company secured a new drug candidate for epilepsy. The company’s foundation-based governance structure provides the grounds for the execution of such long-term strategies. “Since the Lundbeck Foundation holds approximately 70% of Lundbeck's shares, stable investment is possible from a long-term perspective,” said Ding, adding, “The Lundbeck Foundation itself is a major supporter of neuroscience research worldwide.” Currently, Lundbeck is focusing its resources on three core areas through a focused innovation strategy (Focused Innovator) to enhance capital allocation efficiency while pursuing sustainable and profitable growth in the long term. Ding explained, “Lundbeck is focusing on continuing development based on its long legacy in the field of mental illness, strengthening its position in the field of neurology, and expanding into the field of rare neurological diseases. We are concentrating on the acquisition of Longboard Pharmaceutical, the expansion of indications for Vyepti, and next-generation neurological drug programs such as PACAP inhibitors.” Lundbeck is also actively exploring external partnerships to enhance its global competitiveness in the rare disease field. Ding said, “Recently, we have been actively exploring external assets and technologies such as artificial intelligence (AI), neuroimmunology, and RNA-based technologies. When collaborating with external institutions, Lundbeck places importance on scientific excellence, alignment with our strategic focus areas, and the potential to deliver meaningful medical outcomes.” Lundbeck Asia, including South Korea, is also one of the regions that Lundbeck is focusing on. Lundbeck signed a contract with AprilBio in South Korea in October 2021 to introduce the technology for APB-A1, a candidate drug for autoimmune diseases, worth USD 448 million (approximately KRW 537 billion). The contract includes a non-refundable upfront payment of USD 16 million (approximately KRW 19 billion) and milestone payments of up to USD 430 million (approximately KRW 518 billion) based on the progress of development. Ding stated, “The agreement with AprilBio marks a strategic milestone for Lundbeck in accelerating its expansion into the neuroimmunology field. The program aligns well with Lundbeck's R&D strategy that focuses on biomarker-based, traceable development.” She added, “APB-A1 is a promising candidate with potential efficacy in various neuroimmunological diseases and is currently in Phase Ib clinical trials for thyroid eye disease.” Lundbeck's mission is to “advance brain health and transform lives.” Ding mentioned that the company always adheres to this management philosophy as the principle of all its strategies. Ding said, “Lundbeck is committed to scientific innovation that pushes the boundaries of neuroscience, with a patient-centered mindset at the core of all decision-making. We prioritize ethical and responsible research, placing the highest value on safety, transparency, and scientific integrity.” “Lundbeck is actively engaging with the outside world based on its philosophy of focusing on long-term impact rather than short-term results and working with various stakeholders to achieve greater progress. This principle is in line with Lundbeck's core values of passion, responsibility, and commitment to progress.”
- Company
- Samsung Medison aims to achieve 'competitive edge in tech'
- by Whang, byung-woo Jul 22, 2025 06:09am
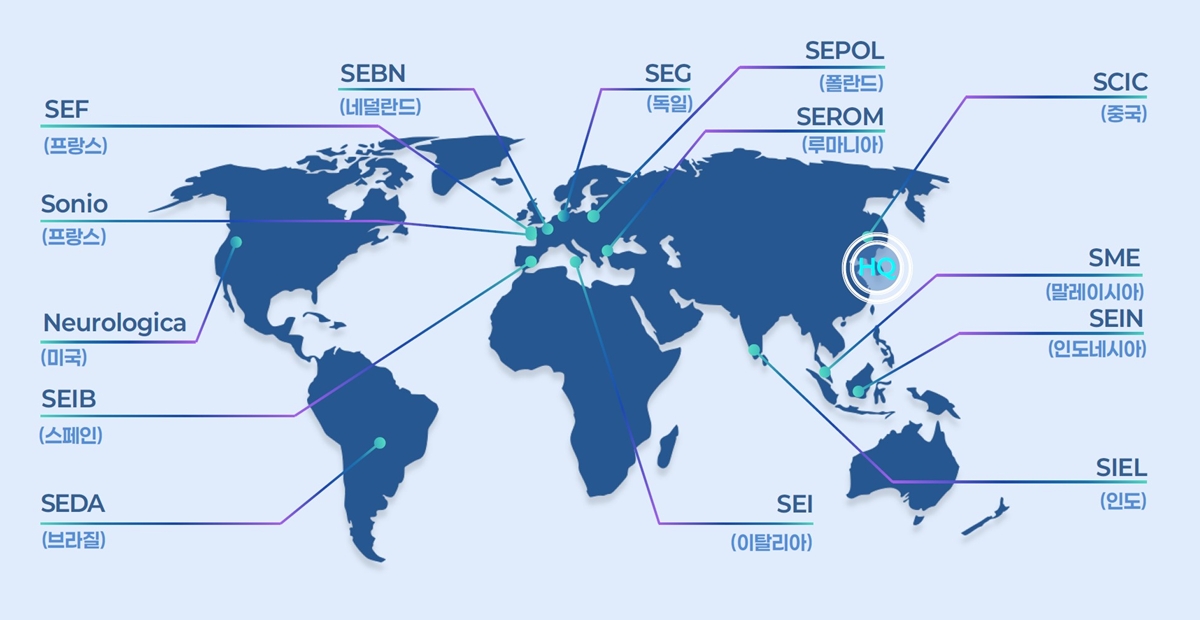
- Samsung Medison, a leading company in South Korea's medical device industry, is celebrating its 40th anniversary this year by investing heavily to become a top player in the diagnostic ultrasound equipment sector. Over the past 40 years, Samsung Medison has grown into a global medical device company, supplying diagnostic ultrasound equipment to over 100 countries worldwide and achieving more than 90% of its revenue through exports. Since Samsung Group acquired this company in 2011, Samsung Medison has consistently strengthened its product competitiveness through technology development, including improvements in imaging performance, the development of AI-powered diagnostic assistance features, and the implementation of user-centric designs. As a result, Samsung Medison showed over 14% growth in the first half of this year compared to the same period last year, and it is anticipated to record its all-time highest annual sales. Strong growth in the first half was driven by winning public tenders in multiple regions, including Europe, and expanding its presence in large hospitals across the U.S. Samsung Medison plans to take a leadership position in the diagnostic ultrasound equipment industry through comprehensive investments in future growth, including ▲Strengthening global sales capabilities ▲Securing next-generation technologies like AI ▲Expanding its portfolio and production capacity. To sustain its high growth in the second half of the year, Samsung Medison plans to recruit top talent, primarily in the U.S. and Europe, to enhance its direct sales capabilities. It will also subdivide and establish specialized sales organizations by medical division across different regions to boost its responsiveness to local markets. Additionally, the company aims to accelerate revenue growth by exploring new large markets, including Australia, Canada, and Mexico. Samsung Medison Global Sectors: France, Netherlands, Germany, Poland, Romania, China, Malaysia, Indonesia, India, Italy, Brazil, Spain, United States. The company also plans to focus on securing next-generation technologies, such as AI, which have recently become a hot topic, to ensure growth through maintaining a 'competitive edge in technology.' Currently, Samsung Medison is establishing an R&D system focused on AI capabilities and actively pursuing strategic partnerships with leading global medical institutions. It plans to expand the utilization of infrastructure, such as Samsung Electronics' overseas research institutes, and establish Innovation Labs and Next Generation Tech Labs locally. These efforts will drive the development of next-generation ultrasound technologies and diagnostic solutions while accelerating new market penetration. Furthermore, Samsung Medison will actively expand its portfolio beyond obstetrics and gynecology and radiology to include divisions such as cardiology·point-of-care diagnostics. The company aims to increase its global market share by strengthening its product lineup with AI features that automatically visualize·quantify the size of major organs and disease indicators in real time across various divisions, such as the heart, liver, and uterus. In line with increasing global demand, the company is also actively pursuing the expansion of its production facilities. The Hongcheon plant, currently undergoing expansion, will incorporate automation systems and smart processes, with plans to increase annual production capacity by over 50% by 2026. By 2030, when the expansion of the second plant is complete, the company aims to more than double its current production while also further enhancing quality. Kyu Tae Yoo, CEO of Samsung Medison, said, "Samsung Medison will continue to develop next-generation medical technologies to provide better diagnostic experiences for both medical professionals and patients." Yoo added, "We will combine the trust built with global medical professionals over the past 40 years with proactive investment to be reborn as a leading global diagnostic device company."

- Company
- Astellas Korea's Seonhee Lee appointed GM of Astellas Egypt
- by Whang, byung-woo Jul 22, 2025 06:07am
- Seonhee Lee, Commercial Excellence & Market Access Head at Astellas Pharma Astellas Korea announced on July 21 that the company has appointed Seonhee Lee, currently a Commercial Excellence & Market Access Head at Astellas Pharma, as the new General Manager of Astellas Egypt, effective from August 1.. Lee has 23 years of experience working in major pharmaceutical companies in South Korea and overseas and is a healthcare expert. She will be the first from the Korean pharmaceutical industry to step up as the GM of Astellas in the Middle East region. The current appointment is regarded as Astellas' strategy to seek and foster talents based on diversity and global career opportunities. Lee, who joined Astellas Korea in March 2024, has been leading the Commercial Excellence team and achieved various successes, including establishing the strategy, strengthening capacity, and advancing omnichannel marketing. After that, she expanded her leadership to the Market Access area, carefully establishing and conducting an insurance reimbursement listing strategy for anticancer agents. Lee has significantly contributed to increasing the likelihood of the market entry for Astellas' core pipeline products. Previously, Lee served in the key leadership positions, including Business Unit Head, Market Access, and global market, in various divisions such as vaccine, chronic diseases, specialty, and anticancer agents, while working at MSD Korea, Merck Serono, and JW Pharmaceutical. Astellas recently established its T-MEA (Turkey, Middle East, and Africa) Headquarters in Istanbul, integrating these regions, and is operating to address unmet medical needs in the area. Astellas Egypt was established in 2022 and is actively involved in various therapeutic areas, aiming to expand local treatment options in Egypt, a key country in the T-MEA region. Junil Kim, Head of Astellas Pharma Korea, stated, "Lee's advancement to the global branch is a significant outcome as it symbolizes the recognition of talent of Astellas Pharma Korea globally." Kim added, "Egypt is a major hub in the Middle East and Africa, a region with significant unmet medical needs, so we expect Lee to achieve meaningful results based on her experience and leadership." Meanwhile, Astellas Pharma Korea has been consistently recognized for its capabilities on the global stage, with a series of promotions and appointments of its executives and employees to overseas branches and new positions since last year. Last year, Sumi Moon, Oncology Brand Lead, moved to the Astellas International Region, and Kyungah Park, Medical Affairs Department Lead, was reassigned to the International Region and the China Medical Affairs Department.

- Company
- Original Prolia will remain ultimate despite patent expiry
- by Whang, byung-woo Jul 22, 2025 06:07am
- As an innovative new drug that transformed the treatment paradigm for osteoporosis, Prolia reached its peak last year with record-high sales. However, with its patent expiration in March this year and the looming entry of biosimilars, the market landscape is expected to undergo significant changes. Amgen Korea plans to solidify Prolia's position by responding to these challenges with a strategy that emphasizes “originality” and its accumulated evidence and brand value as the original drug. Hansam Jung, Bone Marketing Manager of General Medicine at Amgen Korea, who oversees the marketing of Prolia and Evenity, expressed confidence that Prolia would remain a leading example of how original brands can maintain their market position even after patent expiry by reinforcing their brand originality. ‘First-in-Class’ Prolia…targets the market equipped with its long-term data Prolia (denosumab), which was approved in Korea in 2014 and officially launched at the end of 2016, has been rapidly expanding its influence in the field of osteoporosis treatment. [BPM] Hansam Jung, Bone Marketing Manager, General Medicine BU, Amgen Korea Initially reimbursed only as a second-line treatment for osteoporosis in 2017, Prolia saw explosive growth after its reimbursement coverage was expanded to include first-line therapy in April 2019. In fact, Prolia’s domestic sales, which stood at approximately KRW 7.5 billion in 2018 just before the reimbursement expansion, surged to around KRW 44 billion in 2019 and exceeded KRW 80 billion in 2020. Since then, the product has continued to post double-digit growth annually. A key driver behind this growth has been Prolia’s robust clinical data. As a first-in-class drug that inhibits bone resorption, Prolia significantly improved patient adherence compared to oral therapies, thanks to its convenient dosing schedule of one injection every six months. It is also the only osteoporosis treatment that has been proven effective and safe for 10 years through a large-scale, long-term clinical trial. Jung said, “In the 3-year FREEDOM clinical trial and subsequent extension studies in postmenopausal osteoporosis patients, the Prolia group showed a 21.7% increase in vertebral bone density and maintained a non-vertebral fracture incidence rate less than 2%, confirming sustained improvements in bone density and reduced fracture risk through 10 years.” In particular, the risk of spinal fractures decreased significantly from the first year of Prolia treatment, falling 68% lower than the placebo group, while the risk of hip fractures also decreased by more than 40%. According to Jung, Prolia has gained trust as a “treatment that maintains its efficacy and safety even with long-term administration,” establishing itself as the standard first-line treatment for osteoporosis based on such data. “With the addition of real-world prescription experience to the accumulated long-term clinical research data, Prolia is increasingly recognized as a treatment that offers both fracture risk reduction and safety,” said Jung. “We frequently receive positive feedback on its impressive efficacy in improving bone mineral density and its favorable safety profile.” Prolia biosimilars enter the market... Company seeks a way forward by strengthening its marketing capabilities Another factor driving Prolia's growth is its meticulous marketing strategy. Amgen Korea joined forces with Chong Kun Dang from the early stages of Prolia's launch to target the Korean market. Through a partnership formed in 2017, Amgen Korea and Chong Kun Dang divided their cooperation according to their respective strengths. Amgen focused on university hospitals, while Chong Kun Dang targeted smaller hospitals and clinics, ensuring that healthcare professionals across various channels received a consistent message about Prolia's benefits. Jung explained, “Osteoporosis treatments are prescribed in various departments, so we were able to maximize synergy through collaboration with Chong Kund Dang and leveraging its extensive sales network. As a result, Prolia has expanded its prescription range from university hospitals to local clinics and is now the brand with the highest number of patients and sales in local clinics.” Pic of Prolia Amid such strong sales, the market's attention is focused on whether Prolia will be able to keep its lead even after patent expiry. At the end of March, as soon as Prolia's domestic compound substance patent expired, a cheaper biosimilar was listed for reimbursement. As a result, the price of Prolia was adjusted to KRW 123,760, a 20% reduction, from April 1. Although such price cuts will directly impact the original drug Prolia’s sales, some observers note that the growing population of osteoporosis patients due to an aging population could offset this decline through market expansion. Jung said, “As we enter a super-aged society, the number of patients requiring Prolia treatment will continue to increase in the future. While there may be a temporary decline in sales due to the price reduction, we expect to maintain growth by increasing the number of treated patients.” In fact, Prolia achieved sales of approximately KRW 170 billion in South Korea last year, reaching its growth peak. Amgen Korea plans to continue the steady growth of Prolia based on solid clinical evidence and its power as the original brand. Jung explained, “In the face of biosimilar launches, we will further highlight Prolia's strong efficacy and 9 years of real-world experience to emphasize its originality. Prolia can be prescribed for osteoporosis treatment across various medical departments, and we believe that our partnership with Chong Kun Dang will maximize synergy.” Will seek dual brand market penetration with Prolia and Evenity Meanwhile, Amgen Korea is pursuing a market strategy centered on its ‘Bone Portfolio’ for osteoporosis, leveraging a dual-brand approach with Prolia and Evenity to strengthen its presence in the osteoporosis treatment market. Evenity (romosozumab) is the world’s first anabolic agent that combines two mechanisms of action—inhibiting bone resorption and promoting bone formation. Introduced in Korea at the end of 2019, it has since established itself as a first-line treatment for patients at very high risk of fracture. Jung stated, “Prolia and Evenity are key strategic assets unique to Amgen. While their roles differ, we can deliver optimal treatment benefits to a broader range of patients by leveraging their synergy through sequential therapy and other approaches.” In fact, continuing maintenance therapy with Prolia after 12 months of treatment with Evenity has been shown to provide additional bone density improvement and fracture risk reduction, leading to the active adoption of the “Evenity-Prolia sequential treatment approach” regimen in practice. Finally, when asked to describe Prolia in one sentence, Jung replied, “The ultimate osteoporosis treatment for a fracture-free future.” This means that it is a treatment that can help osteoporosis patients achieve a “fracture-free life” through its long-term fracture risk reduction effects. He added, “Osteoporosis is a chronic disease requiring long-term treatment, and at the core of such sustained therapy is Prolia, which has proven its long-term efficacy through data. We will focus on creating success stories for Prolia to emphasize its originality further and ensure that more osteoporosis patients can benefit from its treatment.”

- Company
- Merck Life Science-KAIST hold workshop to share achievements
- by Whang, byung-woo Jul 22, 2025 06:06am
- Merck Life Science-KAIST Workshop Merck Life Science Korea announced on the 21st that it held the “Merck-KAIST Workshop” in Boston, USA, for two days from July 17th to 18th in collaboration with KAIST. The workshop marked the first anniversary of the strategic partnership signed between Merck Life Science and KAIST in May last year, and served as a venue to share achievements and discuss future directions for joint research and collaboration. At the workshop, Merck highlighted several key accomplishments from the past year, including: ▲ the establishment of a new model for academia-industry collaboration; ▲ practical research to address real-world industrial challenges; and ▲ contribution to the life science R&D ecosystem and talent pipeline in Korea and Daejeon City. In addition, the two sides discussed innovative strategies for AI-based new material development and lab automation, exchanged in-depth opinions on leading-edge technology development, and explored future directions for cooperation at the event. The first day of the workshop began with the 2026 Merck Research Challenges session, where Merck introduced key themes, strategic directions, and participation opportunities for its ongoing collaborative research initiatives. Participants then visited the Merck M Lab in Burlington, followed by a Steering Committee meeting to review the progress of key activities, including ongoing research projects within the scope of the partnership, and to discuss outcomes and future directions. The second day featured an Open Innovation Exchange session and the Merck Fellow Award ceremony. The award ceremony was held for the first time this year, and three KAIST professors were selected as recipients based on criteria such as the alignment with Merck's innovation strategy and research, technical excellence, and the commercialization potential of their research. Karen Madden, Chief Technology Officer of Merck Life Science, stated, “The strategic partnership between Merck Life Science and KAIST has evolved beyond collaborative research, developing into a global model of academia-industry cooperation in areas such as R&D and talent cultivation. Through this workshop, we hope to further deepen the partnership between the two institutions and create more tangible research and innovative collaboration outcomes.” Kwang Hyung Lee, President of KAIST, remarked, “KAIST is expanding its unique scientific and technological capabilities globally through its partnership with Merck Life Science, a leading global science and technology company. We look forward to meaningful research outcomes in the field of life sciences through various research collaborations and talent exchanges in the future.” Meanwhile, Merck Life Science and KAIST have been generating diverse collaborative outcomes across life sciences and materials science since signing a memorandum of understanding in May 2024. They are actively conducting joint research to address industrial challenges across a wide range of innovation fields, including next-generation life sciences, new modalities, AI and digital technologies, sustainability, and future-oriented laboratories and facilities.

- Company
- Minimally invasive 'da Vinci robotic surgery'
- by Whang, byung-woo Jul 22, 2025 06:05am
- As robotic surgery becomes a global standard, the industrial robotics market continues to grow in size. In South Korea, where Intuitive Surgical's first Asian branch was established, the market presence of da Vinci robotic surgery is increasingly broadening as it marks its 20th anniversary since its introduction to the country. Intuitive Surgical Korea (hereinafter referred to as Intuitive) recently held a press conference to share its achievements over the past 20 years and its future vision. Da Vinci Robotic Surgical System-Assisted Surgery Synergy with Korean Medical Professionals' Leadership Since the introduction of the da Vinci Robotic surgical systems (hereinafter referred to as 'da Vinci') to South Korea, Intuitive has conducted a cumulative incidence of robotic surgery of over 370,000 cases until now. As of 2024, this record indicates that a surgery was conducted every 8 minute 15 second in average. Yong-Bum Choi, CEO of Intuitive Surgical KoreaKorea is performing single-port robotic surgery (da Vinci), which makes only one incision, more actively than any other country globally, demonstrating the leadership of Korean medical professionals. Indeed, Intuitive has consistently introduced innovative technologies to the Korean market, such as being the first country outside of the U.S. headquarters to launch the next-generation robotic surgery system, 'da Vinci 5 (dV 5),' last year. Yong-Bum Choi, CEO of Intuitive Surgical Korea, said, "Over the past 20 years, robotic surgery has drawn a new standard for surgery amidst technological advancements and changes in the medical environment." Choi added, "This is the result of Intuitive's philosophy of 'Patient First, Always' combined with the efforts of domestic medical professionals." This exceptional surgical skill and leadership of Korean medical professionals are factors that led Intuitive's headquarters to choose Korea as a key innovation hub, enabling close cooperation, from the establishment of its first Asian branch (in 2012) to the prioritized introduction of its latest models. Globally, approximately 17 million surgeries have been performed using Intuitive's da Vinci over the past 30 years. Of this, Korea's accumulated experience accounts for 370,000 cases, playing a significant role in establishing the global standard. Over 43,000 research papers utilizing Intuitive's technology have been published since 1998, with more than 4,000 in 2024 alone, indicating a significant contribution by Korean medical professionals to the dissemination of knowledge. Choi presented his vision, stating, "Just as we turned imagination into reality 20 years ago when robotic surgery first emerged, the next 20 years will see a future where, fused with digital technology, patients can receive the best standard of care anytime, anywhere." Infographic of Intuitive During the press conference, the latest robotic surgery system, da Vinci 5 (dV5), was highlighted. da Vinci 5 features over 150 improvements compared to previous models, notably including a 'Force Feedback' function that allows surgeons to feel the force applied to tissue during surgery with their fingertips, a higher-resolution 3D vision system, and AI-based surgical data analysis capabilities. Hyo Jung Kang, Team Leader at Intuitive Surgical Korea, said, "da Vinci 5 sets a new, higher standard for providing better surgeries to more patients through data utilization." Kang emphasized, "You can experience various functions that simultaneously enhance operating room efficiency and patient safety." Robotic Surgery Has Elevated Surgical Outcomes…Accessibility Expansion remains a Challenge As robotic surgery becomes established in clinical settings, its medical value is recognized for improving patient outcomes, and its societal value lies in elevating and standardizing surgical outcomes. Evidence from clinical research indicates that robot-assisted surgery significantly improves patient prognosis compared to traditional open surgery or laparoscopy. A meta-analysis of over 230 papers published across 22 countries over 12 years for 7 cancer types showed that robotic surgery demonstrated improvements in key indicators compared to other surgical methods, including conversion rates (rate of conversion to open surgery), blood loss, complication rates, length of hospital stay, and readmission rates. Choi stated, "What is important is not the advancement of the technology itself, but that it ultimately allows patients to be freer from complications and pain, benefit from reduced hospital stays, and return to daily life quickly," and added, "In that regard, robotic surgery significantly contributes to social value by elevating and standardizing surgical outcomes." However, broadening the accessibility of robotic surgery was mentioned as a challenge that needs to be addressed. This is because most robotic surgeries in Korea are currently performed on a non-reimbursed basis, leading to a substantial cost burden for individual patients. Regarding this, Choi stated, "I believe no one would oppose the aim of increasing patient access to treatment. If more patients can benefit from robotic surgery through national health insurance coverage, I would certainly support it," and added, "However, as reimbursement is an issue that requires coordination among various stakeholders ,including the government and the medical community, a careful consultation process will be necessary." Intuitive Choi noted that robotic surgery is becoming the standard of care globally. He explained, "Currently, in Japan, robotic surgery is reimbursed for most cancer types, and in Taiwan, 46 additional surgeries were added last year, with a total of 65 surgeries now reimbursed." He found it interesting that "the UK's National Health Service (NHS) recently announced its plan to convert 90% of surgeries currently performed by laparoscopy to robotic surgery by 2035." Along with this, regarding market entry by competitors, Choi said, "While over 60 global companies are developing robotic surgery technology in various ways, Intuitive is confident in maintaining its competitiveness with its accumulated experience and sophiscated technology." Finally, Choi added, "It is rewarding when I hear stories from medical professionals who, during their rounds the day after surgery, personally witness patients who underwent robotic surgery quickly returning to their daily lives." He concluded, "Rapid patient recovery indicates that the value of robotic surgery can be shared with more people."

- Company
- LEO Pharma’s 117-year, single-focus strategy
- by Cha, Jihyun Jul 21, 2025 06:08am
- LEO Pharma is a representative symbol of the success of the Danish biotech industry. It is the oldest pharmaceutical company in Denmark, celebrating its 117th anniversary this year. Originally founded as a pharmacy in Copenhagen, Denmark, the company has grown based on its philosophy of standardizing the quality of medicines and supplying them to everyone. LEO Pharma is recognized as a world-class expert in the development of topical medications such as creams, ointments, gels, and foams that are applied directly to the skin. The wound treatment drug Fucidin, sold by Dong Wha Pharmaceutical in Korea, is LEO Pharma's flagship product. The company has also built a solid portfolio of skin disease treatments, including the psoriasis treatments Enstilum Foam and Xamiol, the non-steroidal atopic dermatitis treatment Protopic, and the eczema treatment Advantan. The reason LEO Pharma has been able to establish itself as a globally respected specialty pharmaceutical company lies in its long-term strategy of selection and concentration. Leveraging its expertise in topical formulations for dermatological conditions, LEO Pharma has remained focused on the field of medical dermatology. This sets it apart from many other companies that prioritize diversifying their drug pipelines and generating quick profits. How has LEO Pharma been able to survive in the global market for over 100 years by pursuing a “single-focus strategy” on a single disease group? Anne Jensen, Vice President of Strategy at LEO Pharma’s headquarters in Ballerup, Denmark, explained that the answer lies in Denmark's unique foundation-centered governance structure. LEO Foundation is the largest shareholder of LEO Pharma with approximately 80% of the shares. The remaining 20% is held by Swedish private equity fund Nordic Capital. Jensen explained that the company was able to consistently invest in specialized areas such as dermatological treatments — which are often difficult to profit from in the short term — because it was shielded from short-term performance pressures and external investor influence. Under the control of the Leo Foundation, which values sustainability over short-term profits, LEO Pharma was able to boldly introduce external innovations and invest in technology. LEO Pharma entered the biopharmaceutical market in earnest in 2016 after acquiring tralokinumab, a candidate drug for the treatment of moderate-to-severe atopic dermatitis from AstraZeneca. LEO Pharma aims to continue building a sustainable growth foundation in the medical dermatology market by pursuing a strategy of securing both internal and external technologies. Anne Jensen, Vice President of Strategy at LEO Pharma - What factors have contributed to LEO Pharma's growth into a global pharmaceutical company specializing in dermatology? LEO Pharma's growth over the past few years has been driven by the success of its atopic dermatitis treatment, Adtralza. This medication, which contains the active ingredient tralokinumab and works by inhibiting IL-13, has significantly strengthened the company’s position in the medical dermatology field. Moving forward, the company plans to drive future growth by leveraging ‘Anzupgo,’ a topical JAK inhibitor for the treatment of chronic hand eczema. Anzupgo is currently available in seven European countries and is awaiting approval under the Prescription Drug User Fee Act (PDUFA) in the United States. - What are LEO Pharma's priorities in terms of expanding its current treatment areas and pipeline? LEO Pharma has established itself as a global leader in medical dermatology. The company is currently focusing on diseases with high unmet medical needs and is pursuing a strategy of jointly developing innovative technologies with partners. Our goal is to leverage the company’s development and commercialization capabilities after introducing new technologies to create maximum value for both patients and companies. For example, we recently began clinical trials to expand the indications for Anzupgo to include palmoplantar pustulosis (PPP) in addition to chronic hand eczema. -What is your outlook for the future of immunology-based skin disease treatments? Over the past 10 to 15 years, the key change in skin disease treatment has been the introduction of immunotherapies. Biologics such as Adtralza, developed by LEO Pharma, have opened up new horizons in the treatment of skin diseases. In the past, skin diseases were considered relatively minor conditions, but now their impact on quality of life is being properly recognized. Skin diseases include more than 1,000 disease groups, and more than 90% of them have no approved treatments. With the advancement of biologics, we predict that the expansion into new disease groups such as atopic dermatitis, psoriasis, and chronic urticaria will accelerate. Our company believes that it can maintain its differentiated positioning based on its agility as a mid-sized pharmaceutical company and its expertise in dermatology by focusing on neglected disease groups that are not addressed by big pharma. -What is LEO Pharma's core strategy in terms of open innovation? We aim to create a “network-based open innovation model.” This strategy involves introducing or jointly developing external assets that have the potential to generate the highest profits for LEO Pharma and provide benefits to patients. From our perspective, this is why it is essential to collaborate with various stakeholders, including academia, patient groups, biotech companies, and pharmaceutical companies. Last year alone, we signed one collaboration agreement each with the Parker Institute and Debra Research, which we expect to play an important role in LEO Pharma's growth. The joint development agreement for STAT6 inhibitors that we signed with Gilead is one of the largest preclinical deals in the industry, with an upfront payment of USD 250 million and a total contract value worth USD 1.7 billion. -What are your thoughts on the Asia-Pacific market that includes South Korea? We are very positive about South Korea and the Asia-Pacific market. South Korea has world-class biotechnology, research capabilities, and an innovative ecosystem, providing an ideal environment for partnerships. We have been covering Korea’s market through excellent distribution partners in South Korea. From a broader perspective of the Asian market, we believe that there are significant opportunities in this region as demand for advanced skin disease treatments is increasing. In fact, China is LEO Pharma's second-largest partner, and we also have a strong local presence in Japan. LEO Pharma plans to further strengthen its position in Asia. - Are there any plans to collaborate with Korean biotech companies, hospitals, or academia? Although we cannot disclose any specific details at this stage, LEO Pharma is always open to opportunities for collaboration with leading Korean companies, hospitals, and institutions. Given Korea's strong reputation in innovation and biotechnology, we believe there is great potential for collaboration. - Does LEO Pharma have any principles that it adheres to in relation to new drug development? Many of the more than 1,000 skin diseases still lack adequate treatment. We believe that innovation requires collaboration, and we are committed to building partnerships with pharmaceutical companies, biotechnology companies, academia, the medical community, and patient organizations. In particular, LEO Pharma is seeking solutions for patients with skin diseases around the world in collaboration with various partners based on five core values: integrity, customer focus, innovation, passion, and adaptability.
- Company
- Omjjara may be prescribed at general hospitals in KOR
- by Eo, Yun-Ho Jul 21, 2025 06:07am
- The new myelofibrosis drug ‘Omjjara’ can now be prescribed in general hospitals in Korea. According to industry sources, GSK Korea's myelofibrosis treatment Omjjara (momelotinib) recently passed review by Drug Committees (DCs) of 13 medical institutions, including Samsung Medical Center, Seoul St. Mary's Hospital, Asan Medical Center Seoul, Kyungpook National University Hospital, Dong-A University Hospital, Seoul National University Bundang Hospital, Ulsan University Hospital, Eunpyeong St. Mary's Hospital, and Ewha Womans University Mokdong Hospital. With the drug’s insurance reimbursement listing in progress, the drug is expected to quickly lead to actual prescriptions once listed. Omjjara passed the Health Insurance Review and Assessment Service's Cancer Review Committee review in March and is currently awaiting submission to the Drug Reimbursement Evaluation Committee. It remains to be seen whether the HIRA process will be completed by the end of this year. The indication currently under review is for the “treatment of myelofibrosis in intermediate- or high-risk adults with anemia.” Omjjara has a triple mechanism of action – it blocks 3 key signaling pathways, not only JAK1 and JAK2 that were inhibited by existing therapies, but also the ACVR1 (activin A receptor type). In the treatment of myelofibrosis, inhibition of JAK1 and JAK2 may contribute to improving systemic symptoms and reducing splenomegaly in patients, while inhibition of ACVR1 may help alleviate anemia by inducing a reduction in hepcidin expression. Anemia management is one of the unmet needs that remain in the treatment of existing myelofibrosis patients. Anemia, which increases blood transfusion dependency, is not merely an issue of dizziness as commonly perceived, but can lead to a life-threatening condition depending on its severity. Omjjara demonstrated significant improvements in key symptoms such as splenomegaly and a reduction in transfusion dependency in patients with anemia-associated myelofibrosis, regardless of prior JAK inhibitor treatment history, in the Phase III SIMPLIFY-1 study and the MOMENTUM study. In the SIMPLIFY-1 study, which evaluated the clinical efficacy and safety of Omjjara in comparison with Jakavi (ruxolitinib) in patients with myelofibrosis and had no prior JAK inhibitor treatment experience, Omjjara demonstrated non-inferiority to ruxolitinib in the primary endpoint of spleen volume response at 24 weeks of treatment. The proportion of patients in each arm who were transfusion-free was 66.5% in the Omjjara arm and 49.3% in the ruxolitinib arm, with significantly lower transfusion dependence (better transfusion independence) in the Omjjara arm. Seo-Yeon Ahn, Professor of Hematology at Chonnam National University Hwasun Hospital, said, “While JAK inhibitors previously used in the treatment of myelofibrosis demonstrated efficacy in reducing splenomegaly and alleviating systemic symptoms, they also posed unmet needs such as worsening anemia or increasing blood transfusion dependency. Omjjara has demonstrated significant clinical value in managing anemia, which is closely linked to the prognosis of patients with myelofibrosis. With its domestic launch, it is expected to contribute to improving treatment outcomes and quality of life for more patients."