- LOGIN
- MemberShip
- 2026-06-22 21:56:21
- Policy
- COVID-19 drugs to be reimbursable, "KRW 50,000 co-payment"
- by Lee, Jeong-Hwan Sep 27, 2024 05:53am
- The National Health Insurance will cover two COVID-19 treatments, Paxlovid and Veklury, starting on October 1st. The government will list COVID-19 treatments for reimbursement and make a policy revision to support the patient co-payment at the previous price of KRW 50,000. The National Health Insurance coverage for Takeda Pharmaceuticals Korea's Zejula Cap (ingredient: niraparib tosylate monohydrate), a treatment for advanced ovarian cancer, will be expended, and the ceiling price of the drug will be reduced, starting on October 1st. Patients with advanced ovarian cancer who paid KRW 41 million in annual treatment costs per person are expected to pay KRW 2..05 million in annual treatment costs with the National Health Insurance coverage. On September 26th, the Ministry of Health and Welfare (MOHW) held the 19th Health Insurance Policy Review Committee meeting and decided on items related to National Health Insurance coverage of COVID-19 treatments and expanded National Health Insurance coverage for Zejula Cap. COVID-19 drugs will be covered by NHI…expanded reimbursement for Zejula The two COVID-19 treatments that the Korea Disease Control and Prevention Agency (KDCA) purchased and supplied will be covered by the National Health Insurance, starting in October. Those two are Paxlovid and Veklury. The ceiling price for Pfizer Korea's 'Paxlovid' is KRW 941,940 per 30 tablets in a package, and that for Gilead Sciences Korea's 'Veklury 100mg Powder For Concentrate For Solution For Infusion' is KRW 520,000 per vial. The related policy will be revised along with the reimbursement listing of these drugs. As an administrative action to prevent patient co-payment increases due to reimbursement, the co-payment will be maintained at the current cost of KRW 50,000. Starting next month, the reimbursement coverage for Zejula Cap, a treatment for advanced ovarian cancer, fallopian tube cancer, and primary peritoneal cancer, will be expanded, and its ceiling price will be reduced. For Zejula Cap, the usage criteria will be expanded, and it will be reimbursable for maintenance therapy of patients with advanced epithelial ovarian cancer, fallopian tube cancer, and peritoneal cancer who are responsive to the first-line platinum-based therapy. Previously, it was reimbursable only when the patients tested BRCA mutation-positive ovarian cancer from genetic screening. Starting next month, the drug will also be reimbursable in all homologous recombination deficiency-positive genetic mutations, including ovarian cancer-associated genomic instability identified from genetic testing. Patients with advanced ovarian cancer who paid KRW 41 million in annual treatment costs per person are expected to pay KRW 2..05 million in annual treatment costs with the National Health Insurance coverage. The pilot project for primary healthcare home visits will be improved Since December 2019, the government has been implementing a pilot project for primary healthcare home visits, a healthcare service in which private clinic doctors visit patient homes, to enhance healthcare access for patients who have difficulties visiting medical centers. Starting in November, the designation of medical centers for healthcare home visits will be expanded to include hospitals (local medical centers). Peviously, only private clinics and oriental medicine clinics were eligible for designation. Now, hospitals (local medical centers) can also participate following the policy revision. It will be expanded for patients with severe disease who require home medical care, including bedridden patients with Grade 1·2 long-term care and patients with severe disease using medical devices (oxygen therapy and a respirator). Co-patients for home visits will be reduced for these patients. In order to implement a reduction in out-of-pocket costs for eligible patients, a medical center's screening process will be developed. Additionally, a computer system for patients to claim the reduction of out-of-pocket costs is also in development. The implementation of the out-of-pocket cost reduction is scheduled to occur after November. In addition, to expand the primary healthcare home visit project, additional participating institutions will be invited in October. Extended NHI support for emergency care The policy for providing the National Health Insurance coverage for emergency care, which costs KRW 208.5 billion per month, will be extended to prevent a gap in healthcare for patients with severe and emergent symptoms amid the doctor's strike. The budget will be used to strengthen compensation for emergency departments and tertiary general hospitals so that they can focus on the treatment of emergency and patients with severe symptoms, and for the return of patients with mild symptoms to hospitals and clinics. Incentives will be increased to ensure the prompt transport of critical patients to hospitals. Additionally, incentives for medical care in emergency centers, including emergency care fees and CPR fees, will be enhanced. To facilitate prompt hospital responses to critical patient emergencies, the government will provide financial support for specialists treating hospitalized critical patients and incentives for critical patient hospitalization. The emergency medical care analysis comparing year-over-year from March to July 2024 shows that hospitals are maintaining critical patient care systems. However, the number of critical patients visiting the regional·local emergency medical centers has slightly decreased. The government will extend additional support to strengthen emergency care treatment capacity. The government will also extend raises for specialist medical fees, which were temporarily implemented during Chuseok to maintain the infrastructure of critical·emergency patient treatment at emergency medical centers. The raises for critical·emergency surgeries will also be extended. The raises in specialist medical fees will be 250% for regional·special emergency medical centers and 150% for local emergency medical centers. The raises for critical·emergency surgeries at regional·special·local emergency medical centers will be 200%. The MOHW stated, "We will provide a temporary insurance fee for emergency medical care to prevent a medical care gap for emergency and critical patients. We aim to promptly solve the current situation to ensure people do not encounter difficulties using medical centers."
- Company
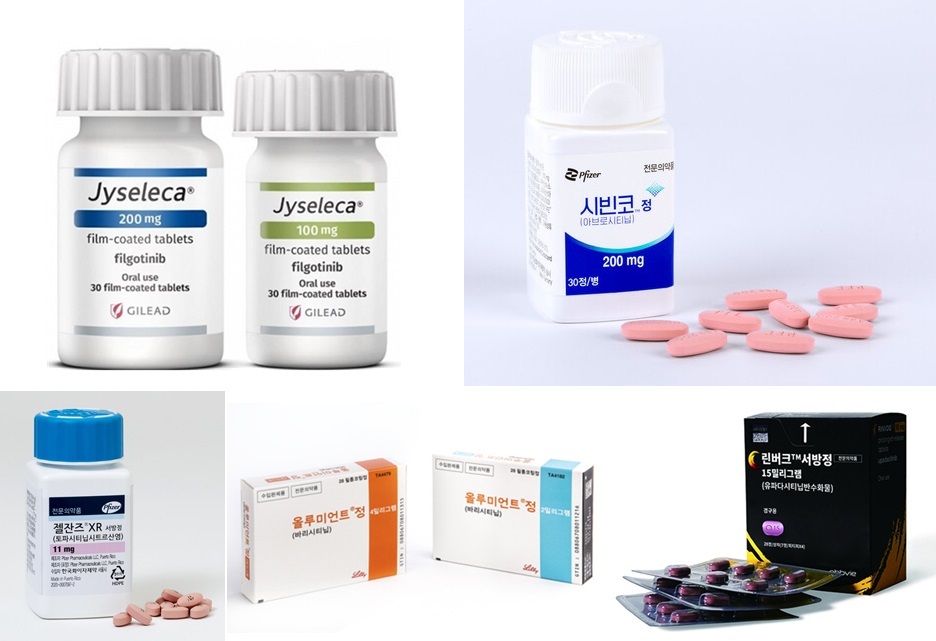
- Switching between JAKis for RA yet to be reimbursed
- by Eo, Yun-Ho Sep 27, 2024 05:52am
- 국내 처방되고 있는 JAK억제제들The plan to allow insurance reimbursement when switching between JAK inhibitors in rheumatoid arthritis, which was expected to take effect in October, has been postponed. According to Dailypharm coverage, the health authorities have recently put on hold the notification of the proposed revision that allows switching between JAK inhibitors in rheumatoid arthritis. The tentative timeline for the notification was set as the end of the year, but this is not confirmed yet. The delay was reportedly caused by problems with the final approval of the budget bill. Currently, 4 drugs are prescribed for rheumatoid arthritis in Korea, including Pfizer Korea’s Xeljanz (tofacitinib), Lilly's Olumiant (baricitinib), AbbVie's Rinvoq (upadacitinib), and Eisai Korea’s Jyseleca (filgotinib). Until now, the government has been adhering to the position that it is difficult to reimburse switching JAK inhibitors due to the lack of clinical evidence. However, after continuous statements submitted by the Korean College of Rheumatology and other organizations, as well as prescription experience on cross-dosing, the government reconsidered its position and came to a positive conclusion. The industry expected the change to be applied in October, but the notification of the amendment has been delayed. As a result, it remains to be seen if patients will be able to freely switch between JAK inhibitors before the end of the year. If reimbursement for switching between the drugs is allowed, such use is expected to further activate the JAK inhibitor market. According to drug market research firm UBIST, the outpatient prescription market for JAK inhibitors was KRW 27.5 billion in the first half of last year. This is a 54% increase in one year compared to the KRW 17.8 billion in the first half of last year. The market for JAK inhibitors has been expanding at a rapid pace. The market, which had been around KRW 12.5 billion in 2019, had expanded to KRW 18.7 billion in 2020, KRW 25.5 billion in 2021, KRW 33.5 billion in 2022, and KRW 40 billion last year. This year, the market reached KRW 27.5 billion in the first half of the year alone and is expected to exceed KRW 50 billion by the end of the year.
- Policy
- 2nd Approval-Evaluation-Negotiation pilot project imminent
- by Kim, Jin-Gu Sep 26, 2024 05:51am
- The initiation of the 2nd pilot project for the ‘approval-evaluation-negotiation linkage system,’ which was introduced to shorten the time from drug approval to reimbursement, is imminent. The Ministry of Health and Welfare has completed receiving drug submissions for the 2nd pilot project from pharmaceutical companies and is in the final review stage. The MOHW plans to implement the 2nd pilot project after reflecting on the improvements identified during the 1st pilot project. Eun-Joo Lee, an official from the Ministry of Health and Welfare’s Division of Pharmaceutical Benefits, announced so regarding the pilot project linking the approval-evaluation-negotiation system at the ‘Roundtable on Improving Access to Rare Disease Drugs' that was held at the National Assembly on the 24th. The government implemented the 1st pilot project for the approval-evaluation-negotiation linkage system last year. The project consists of simultaneous approval by the MFDS, reimbursement evaluation by the Health Insurance Review and Assessment Service, and drug price negotiations by the National Health Insurance Service. The project shortens the 300-day review period required for new drug access in Korea – which consists of an MFDS approval period of 120 days, 150 days of reimbursement evaluation by HIRA, and 60 days of drug price negotiations by the NHIS - and strengthens Korea’s access to new drugs. Two drugs - Qarziba Inj (dinutuximab, Recordati) and Bylvay (odevixibat, Ipsen) – were selected for the 1st pilot project. At the time, the government proposed the following criteria for selection: ▲a drug for cancer or rare diseases with a life expectancy of less than one year; ▲has a small number of patients; ▲lacks alternative drugs; ▲has a survival period over two years and demonstrated a superior treatment effect. Several companies submitted applications to participate in the pilot program, among which the government ultimately selected 2 drugs that met the selection criteria. Although the 1st pilot project has not been completed yet, the Ministry of Health and Welfare has been promoting to launch of the second pilot project. “We have completed receiving applications for the 2nd pilot project. We now need to select the drugs that meet the conditions of the 2nd pilot project,” said Lee. He added, “The number of drugs for the 2nd pilot project has not been decided upon yet. There are no criteria that require us to select only 2 drugs as in the 1st project. We will select drugs that meet the conditions.” The Ministry of Health and Welfare has proposed 4 criteria for selecting drugs for the 2nd pilot project: ▲ drugs that can apply for approval and decision by the end of June next year; ▲ drugs with sufficient effect for the treatment of life-threatening diseases (life expectancy of less than 1 year) or rare diseases; ▲ drugs that have no existing treatment or have shown clinically significant improvements in efficacy over existing treatments; and ▲ drugs that have been designated or are eligible for Global Innovative products on Fast Track (GIFT). To be selected as a drug for the 2nd pilot project, the drug needs to satisfy all the conditions above, and the company needs to submit the following data: ▲estimated financial expenditure per capita and total estimated financial expenditure; ▲results of overseas reimbursement evaluations; ▲the number of listed overseas countries; ▲the name of listed countries; and ▲name of the company. The MOHW plans to analyze the pros and cons of the 1st pilot project and apply them to the 2nd pilot project. In the 1stpilot project, the approval and reimbursement evaluation processes for Qarziba have been completed. The agenda had passed the Drug Reimbursement Evaluation Committee review and is currently being reviewed upon the company's reapplication. Compared to the existing system, the time from its approval to reimbursement evaluation has been significantly reduced. However, in the case of Bylvay, there are some delays due to difficulties in the evaluation process. The marketing authorization was completed on the 23rd of last month, but the reimbursement evaluation process was hampered due to the use of a difficult evaluation tool. However, the government is focusing on shortening the timeframe by conducting pre-negotiations at the same time. The HIRA explained that it will focus on how the companies manage uncertainties during the reimbursement evaluation process in the 2nd pilot project “In the future, uncertainty management will be important in the reimbursement evaluation process,” said Kook Hee Kim, Director of the Pharmaceutical Benefits Management Division at HIRA. ”Since the drugs are expected to be effective but not yet certain, the pharmaceutical companies should discuss how to address the uncertainties and prove their drug’s effect in the post-marketing process after the fact and share the risk with the government.” “The approval-evaluation-negotiation process is not a guarantee that the drug will be reimbursed. It means that the evaluation period is shortened,” explained Kim. ”The companies should submit data on how they will prove the uncertainties in the post-marketing process. Otherwise, the drug’s reimbursement may be delayed.”
- Company
- Minjung Jung to head Corporate Affairs at Sanofi KR, NZ, AU
- by Eo, Yun-Ho Sep 26, 2024 05:51am
- Sanofi’s Executive Director Minjung Jung (48) has been named head of Corporate Affairs for the Sanofi Group's 3 subsidiaries - Korea, New Zealand, and Australia. The appointment follows the appointment of Kyungeun Bae (53) as the General Manager of Pharma MCO South Korea and Australia/New Zealand & MCO Lead in the first half of last year, raising the stature of Sanofi Korea. With this promotion, Jung will now lead Corporate Affairs for the three countries, effective September 9th. The Corporate Affairs department is responsible for Public Affairs, Communications, CSR, Government, and Affairs. Jung is a seasoned public affairs professional who joined Sanofi in September last year. Since starting her career at MSD Korea in 2007, Jung has served various marketing roles at Boehringer Ingelheim Korea, Merck Korea, and Amgen Korea. Sanofi has been executing a Play To Win strategy, which focuses on building innovation platforms to develop first-in or best-in-class therapies and vaccines from 2020. The plan is to drive long-term growth through a virtuous cycle of efficiently redeploying resources to accelerate innovation and maximize R&D productivity. As a key part of this strategy, Sanofi has been operating its Korea and Australia & New Zealand subsidiaries as one integrated organization.

- Opinion
- [Reporter’s View] How to increase the new drug approval fee
- by Whang, byung-woo Sep 26, 2024 05:51am
- The Ministry of Food and Drug Safety (MFDS) decided to significantly raise its new drug approval fee to expand its capability for the prompt review of newly developed drugs. The fee for new drug approval, which was previously KRW 8.83 million, was increased 48 times to KRW 410 million based on the benefit principle. Despite the dramatic increase, there has been little backlash as Korea’s drug approval fee has been relatively low compared to overseas. The MFDS aims to raise the new drug approval fee from next year after a 60-day administrative notice period. As the pharmaceutical industry has been actively supporting the realization of the fee, there have been more expectations than concerns. Rather, the industry's concern is whether the fee hike will have an immediate effect. The MFDS plans to use the increased fees to recruit specialized review personnel and expand the proportion of high-competency reviewers, such as doctors, pharmacists, and those with at least 3 years of postdoctoral experience, from the current 31% to 70%. However, whether the government can secure the required specialized personnel remains in question. While the MFDS emphasizes the importance of securing human resources by allocating half of the fee hike to labor costs, it is unclear at what point it will be able to recruit the desired amount of human resources. This is also a concern for pharmaceutical companies. While there is no doubt that the fee hike will speed up approvals, there is also concern that pharma companies will not immediately see the benefits of their inputs. As a result, some pharmaceutical companies are considering accelerating their submission plans and applying for approval before the fee hike. For example, there are concerns about whether the review process for new drug approvals will change dramatically before and after the fee increase, or whether the new system will take time to settle in. In the end, the pharmaceutical industry needs the MFDS to preemptively establish a system so that the increased fees can be utilized as intended. Apart from the consensus on the KRW 400 million increase in review fees, the system should not have a backward-looking investment structure that raises fees and then uses them to increase the number of reviewers. With the expected improvement in the system is expected, attention is likely to be focused on the first case of their application. In the bigger picture, the MFDS is hoping that the fee hike will also prevent the poke-and-see applications without sufficiently demonstrating safety and efficacy, in addition to shortening the review period. In other words, it could act as a hurdle to prevent companies from using Korea’s regulatory authorities as sort of a consulting window. However, the pharmaceutical industry is also hoping that the MFDS’s review will also include consultation at the approval stage, as the authorities are requesting new drug fees at the level of FDA and EMA. The increase in drug approval fees is expected to bring about many changes in the domestic regulatory environment. As the saying goes, 'the beginning is half the battle', so we expect thorough preparation and changes to be made to meet the purpose of the MFDS’s new drug approval innovation plan.
- Policy
- Higher dosage Eylea expected to strike back at biosimilars
- by Lee, Tak-Sun Sep 26, 2024 05:51am
- The competition in the market for the macular degeneration drug Eylea has recently intensified with Samsung Bioepis and Celltrion, which have recently launched biosimilars. The original developer is set to strike back against the competitors. Bayer will release Eylea Inj 8 mg (aflibercept), with an extended treatment interval, into the market next month at a relatively cheaper price. According to the industry on September 25th, Eylea Inj 8 mg will be listed for reimbursement next month at a price lower than the evaluation price, KRW 795,000. Eylea Inj 8 mg provides a long-lasting, effective dosage within the eye at a four times higher dosage than the previous 2 mg. It extends the treatment interval while reducing the number of injections. Eylea Inj 8 mg is injected once monthly for an initial 3 months. Then, based on visual and anatomical test outcomes, a treatment interval can be extened up to 16 weeks at the physician's judgement. After the initial extension of the treatment interval, it can be extended up to 20 weeks with the Treat-and-Extend(T&E) therapy. This approach allows flexibility in adjusting the treatment interval depending on the patient's condition while maintaining stable vision and anatomical test outcomes. The original and biosimilars currently sold in the market are at a 2 mg dosage. For neovascular age-related macular degeneration (nAMD) treatment, the drugs are injected once monthly for an initial 3 months, followed by a once every 2 months treatment regimen. Using the new 8 mg will extend the treatment interval more than twice, from once every 2 months to once every 4 months. The PULSAR study secured non-inferiority results of the new product by showing that patients treated with Eylea 8 mg at a 12-week interval had the best corrected visual acuity (BCVA) of 6.7 letters on average from the baseline at 48 weeks, those at a 16-week interval had BCVA of 6.2 letters, and those at an 8-week interval had the BCVA of 7.6 letters. However, the drug price is not more than double the original price. Due to the biosimilar release, the current ceiling price for Eylea Inj·Prefilled Syringe is KRW 496,118, a 30% reduction from the highest price. The newly listing Eylea Inj 8 mg costs KRW 795,000. Despite extending the treatment interval more than twice, the price has not doubled because Bayer has set the price lower than the evaluated price. However, the price is double that of biosimilars. Samsung Bioepis' Afilivu Inj 40 mg/ml costs KRW 350,000, and Celltrion's Eydenzelt Inj costs KRW 330,000. Although the price of biosimilars is low, newly diagnosed patients are likely to choose the high-dose original drugs, which require fewer injections due to the extended treatment interval. Hence, Eylea Inj 8 mg will likely be a direct competitor to biosimilars. Eylea is a blockbuster product, recording KRW 96.8 billion in sales last year, based on IQVIA. Due to its marketability, Samsung Bioepis and Celltrion, which have released their products in May and September, respectively, are trying to seize the market by partnering with pharmacists who are specialists in ophthalmology. Industry personnel said, "Patients who are benefiting from the existing Eylea 2 mg are less likely to switch products. However, new patients are likely to choose from biosimilars, with lower cost, and high-dosage original drug, with fewer injections," adding, "An intense competition among overseas original developer and Korean biosimilar developers is expected."
- Policy
- Potential expanded indication for obestiy drug 'Wegovy'
- by Lee, Hye-Kyung Sep 26, 2024 05:51am
- Product photo of Wegovy. A clinical trial for Novo Nordisk Pharma's obesity drug, 'Wegovy (semaglutide),' will be conducted in South Korea for expanded indication. On September 20th, the Ministry of Food and Drug Safety (MFDS) has approved the 'Phase 2 clinical trial to evaluate the efficacy, safety, and dosage testing of NNC0519-0130, subcutaneously administered once-weekly, in comparison to Ozempic (semagluide) 1.0 mg and placebo in overweight or obese patients who have or do not have type 2 diabetes.' The clinical trial will involve 456 individuals worldwide from December 2024 to September 2026. In South Korea, the study will enroll 32 patients and will be conducted at Pusan National University Hospital, Ilsan Hospital, Hallym University Medical Center, Seoul St. Mary's Hospital, Hanyang University Guri Hospital, and Korea University Guro Hospital. Wegovy received domestic marketing authorization in April with indications to ▲aid weight-loss overweight patients who have a Body Mass Index (hereafter referred to as BMI) of 30kg/m2 or higher or those who are overweight with early BMI of 27kg/m2 or higher and below 30kg/m2 and having one or more weight-related accompanying diseases (abnormal glycemia, type 2 diabetes, high blood pressure, dyslipidemia, obstructive sleep apnea, or cardiovascular diseases) ▲reduce risks of major cardiovascular events in overweight or obese patients with early BMI of 27 kg/m² or higher with cardiovascular diseases. Additionally, the company will conduct the Phase 2 clinical trial to add indication to treat chronic kidney disease. Based on Phase 3 SELECT study results, presented by Dr. Katherine Turtle, a professor at the University of Washington School of Medicine, at a recent European Renal Association meeting, Wegovy can reduce the risk of kidney failure and death in patients with chronic kidney disease because of type 2 diabetes. The clinical trial involved 3533 patients with type 2 diabetes and chronic kidney disease. Half of the participants received weekly semaglutide injections for 3 years, and the rest received weekly placebo. Patients treated with semaglutide had a lower possibility of worsening symptoms that require dialysis or kidney transplant by 24% and a lower possibility of death due to major cardiovascular disease, including heart attack, than those treated with placebo by 29%. Patients treated with semaglutide had a 20% lower possibility of death of all causes during the study period. However, investigators studying semaglutide have not found an exact mechanism by which this drug helps the kidney, and the study was limited in that around two-thirds of the participants were white males. Meanwhile, Wegovy's active ingredient, semaglutide, is a 'Glucagon-like protein (GLP)-1' agonist. GLP-1 receptors lower blood pressure and regulate appetites. The basis for Wegovy's approval in South Korea was the STEP study, in which patients treated with Wegovy (1306 patients) had a 14.9% weight loss across 68 weeks compared to the baseline, showing a significant difference from a 2.4% of the placebo group (655 patients). According to Novo Nordisk Pharma's business performance reporting for the first half of 2024, Wegovy generated US$3.07 billion, up 74% year-over-year.
- Product
- Zomig distributor changes from AZ to SK Chemicals
- by Kim JiEun Sep 25, 2024 05:49am
- Product photo of Zomig Tab.The distributor of Zomig Tab, a triptan used in the treatment of migraine, will change from AstraZeneca Korea to SK Chemical. The distribution industry suggests that the change in a distributor could impact the supply and demand chain. AstraZeneca Korea recently sent an official letter to the pharmaceutical wholesaler community of 'termination of a distribution agreement and changes to distributor' of Zomig Tab. The company stated in the official letter that starting on October 1st, due to the sales agreement, Zomig Tab 2.5 mg distributor will change from AstraZeneca Korea to SK Chemicals. Zomig Tab has been in shortage several times. The Ministry of Food and Drug Safety (MFDS) predicted drug shortages three times between last year and early this year. AstraZeneca reported MFDS that such supply shortages were due to laying in supplies and the company requested more supply volume to the manufacturer. The industry suggests that the current supply shortage may be partially due to AstraZeneca Korea's lack of domestic promotion of the drug after selling the global sales rights of Zomig Tab to another company. After officially learning about the Zomig Tab distributor change, the industry confirmed that an anticipated factor may have contributed to the supply shortages. The wholesaler community predicted that the distribution of the drug might be unstable for a while since the order requests have not been processed properly following the announcement related to the Zomig Tab distributor change. A wholesaler said, "Order request for Zomig Tab did not go through today, and we found that there has been a change to the distributor,' adding, 'Even if SK Chemicals take over the sales rights, distribution will be affected for a while. We think that the drug supply will be impacted for at least 15 days."
- Policy
- First generic version of Qudexy XR Cap is approved in KOR
- by Lee, Hye-Kyung Sep 25, 2024 05:49am
- The first generic version of the topiramate-based extended-release epilepsy treatment ‘Qudexy XR Cap’ has been approved in Korea. The Ministry of Food and Drug Safety granted marketing authorization for Intrio Biopharma’s ‘Topimed XR Tab. 50mg’ on the 23rd. Unlike ‘Qudexy XR Cap’ which is an oblong hard capsule formulation, Topimed XR Tab was developed as a circular extended-release film-coated tablet. Intrio Biopharma has been developing Topimed XR Tab since 2022. The original epilepsy drug that contains topiramate was Janssen's ‘Topamax Tab,’ but in 2014, the U.S. generic company Upsher-Smith developed and received U.S. FDA approval for Qudexy XR Cap, which is an extended-release capsule formulation that is not available with Topamax. Qudexy XR Cap is also the topiramate approved in Korea. The drug was also supplied by SK Chemicals in August 2017 and is listed on the KFDA's Green List and protected until January 2034. However, the newly approved Topimed XR Tab is likely to be released as a film-coated tablet formulation, rather than the capsule formulation that is being protected with a patent. This is because no pharmaceutical company has attempted to avoid Qudexy XR Cap’s patent. Topimed XR Tab is indicated ▲ as initial monotherapy in patients 6 years of age and older with partial-onset or primary generalized tonic-clonic seizures and ▲ as adjunctive therapy in children 2 years of age and older and adults with partial seizures, primary generalized tonic-clonic seizures, and seizures associated with Lennox-Gastaut syndrome (LGS). While existing immediate-release topiramate formulations are taken twice daily, this medication is an extended-release formulation that is absorbed into the body more slowly and can be taken once daily. Topiramate-based formulations are the most prescribed ingredient in the KRW 80 billion antiepileptic drug market with a sales volume of KRW 30 billion, and the outpatient prescription value of Qudexy XR Cap was KRW 3.5 billion based on UBIST last year. The drug has been covered through insurance reimbursement in Korea since February 2018.
- Company
- "Bird flu: a new emerging pandemic risk factor"
- by Son, Hyung Min Sep 25, 2024 05:49am
- Jacob Lee, Professor of the Division of Infectious Disease at Hallym University Kangnam Sacred Heart Hospital.The industry is preparing against avian influenza, which can be transmitted from animal to human, as it is predicted to be the next pandemic risk factor. Experts suggest that an improved vaccine technology development and manufacturing system are required ahead of the emerging pandemic following COVID-19. CSL Seqirus Korea held a press conference on September 24th at Hotel President in Seoul to share the recent advances in avian influenza. During the meeting, Jacob Lee, Professor of the Division of Infectious Disease at Hallym University Kangnam Sacred Heart Hospital, emphasized strengthening Korea's response measure strategy for avian influenza. Avian influenza is an infectious respiratory disease that commonly occurs in wild birds. Recently, there have been cases of animal and human infections in addition to poultry and wild birds. In March, a death case was reported in Vietnam related to avian influenza human infection. The highly pathogenic H5N1 strain of avian flu, which emerged from a type of influenza A and is spreading world-wide, has infected over 300 species of birds and over 40 species of animals. 14 cases related to H5N1 human infection transmitted from cows and birds were reported in the United States alone. Several duck farms in South Korea have recently reported H5N1 infection cases. The Korea Disease Control and Prevention Agency (KDCA) is preparing responses to potential influenza pandemic, such as holding a symposium to discuss response plans. Lee said, "Although continual human-to-human infection has not been reported, there are increasing cases of animal-to-human infection. The academics consider avian influenza as the risk factor for causing the next pandemic." And added, "We could use vaccine technologies developed for COVID-19, such as the messenger RNA (mRNA) platform, for influenza. Establishing these platforms can reduce development time once an antigen is chosen." mRNA vaccines are composed of mRNA molecules and lipid nanoparticles surrounding it. mRNA contains a genetic material that can synthesize target proteins, and lipid nanoparticles protect mRNA, working as a transporter to deliver mRNA into the body. Recent studies showed that lipid nanoparticles can function as not only an mRNA transporter but also an 'immune enhancer,' increasing immune responses to vaccines. Lee says that Korea needs to secure vaccines that can enhance immunity since technology is still lacking. CSL Seqirus is known to manufacture vaccines rapidly through its platform. The company can readily shift from manufacturing seasonal vaccine influenza to pandemic influenza vaccine using its global manufacturing network. Moreover, the company manufactures and supplies vaccines for viral variations and zoonotic viruses. Marc Lacey, CSL Seqirus Global Pandemic Head, said, "We have built vaccine technologies through years of vaccine development experiences. For example, MF59 can enhance immune responses with a small antigen dosage. For H5N1 avian influenza variant, it only took 79 days until the vaccine developed. Our company can readily respond to global pandemic influenza." Lee said, "Vaccines that can be produced domestically in South Korea are egg-based and cell-based. Korea needs to develop the mRNA vaccine platform and immune enhancers. If the development is difficult, we must establish a system to introduce vaccines from overseas."