- LOGIN
- MemberShip
- 2026-06-23 02:35:49

- Company
- Challenges in the evolving lung cancer treatment landscape
- by Son, Hyung-Min Aug 28, 2024 05:51am
- The head-to-head battle between the epidermal growth factor receptor (EGFR)-mutated non-small-cell lung cancer drugs (NSCLC) Leclaza and Tagrisso has carried on to competition of each drug's combination therapy regimens. This month, the U.S. Food and Drug Administration (FDA) approved Leclaza+Rybrevant as a first-line treatment for EGFR-positive NSCLC. This is the first time a targeted therapy plus targeted therapy combination has been approved. Tagrisso was approved in Korea and the U.S. this year after confirming its efficacy in combination with platinum-based chemotherapy. While attention is focused on whether combination therapies will find a place in the first-line treatment market for EGFR-mutated NSCLC, there is a consensus that the choice of a first-line treatment should be based on the patient's comprehensive needs, including side effects, frequency of hospital visits, and quality of life. Leclaza+Rybrevant combo improved overall survival compared with Tagrisso monotherapy NSCLC drug Leclaza Yuhan Corp and its partner Janssen have confirmed the efficacy of the combination of the Leclaza+Rybrevant combination therapy. Leclaza is a third-generation tyrosine kinase inhibitor (TKI) that targets exon 19, and exon 21 (L858R) in EGFR-positive NSCLC. Rybrevant is a targeted treatment option that targets the exon 20, MET mutation. The two companies successfully secured FDA approval for Leclaza+Rybrevant with the MARIPOSA Phase III trial. Specifically, the FDA approval was based on clinical results presented by Janssen at the European Society for Medical Oncology Annual Congress (ESMO 2023) last year. The results showed a median PFS of 23.7 months in the Leclaza+Rybrevantcombination arm, which was longer than the 18.5 months in the Leclaza monotherapy arm, and 16.6 months in the Tagrisso monotherapy arm. Leclaza+Rybrevant combination was associated with a 30% lower risk of disease progression and death than Tagrisso monotherapy. The interim OS analysis showed a trend favorable to Leclaza+Rybrevantover Tagrisso monotherapy. PFS2 results showed a 25% lower risk of disease progression or death in the Leclaza+Rybrevant combination arm compared with Tagrisso monotherapy arm. Tagrisso+platinum-based chemotherapy is approved in Korea and the U.S. NSCLC drug TagrissoTagrisso was approved in Korea based on the results of the FLAURA2 clinical trial. In May, the Ministry of Food and Drug Safety approved Tagrisso+platinum-based chemotherapy as a first-line treatment for EGFR-positive non-small-cell lung cancer. Tagrisso is a third-generation TKI developed by AstraZeneca. The Phase III FLAURA2 trial enrolled 557 patients with locally advanced or metastatic NSCLC who had received no prior systemic therapy and were positive for EGFR exon 19 deletion or exon 21 mutation. The study evaluated the efficacy and safety of Tagrisso combination therapy versus Tagrisso monotherapy. Results showed that Tagrisso+platinum-based chemotherapy reduced the risk of disease progression or death by 38% compared to Tagrisso monotherapy. Median investigator-assessed PFS was 25.5 months, an 8.8-month extension compared to 16.7 months with Tagrisso monotherapy. Median PFS by blinded independent central review (BICR) was 29.4 months, compared with 19.9 months in the Tagrisso monotherapy arm. Also, inpatients with the L858R mutation, Tagrisso+platinum-based chemotherapy showed a median PFS of 24.7 months, 10.8 months longer than the 13.9 months achieved in the Tagrisso monotherapy arm. Use of combination therapies boosts efficacy, but managing side effects remains key#EB Both Leclaza and Tagrisso have been granted additional regulatory approvals for their combination therapies, heating competition in the first-line treatment market for EGFR-mutated NSCLC. However, side effects management will be a key issue due to its combined use with new drugs. Rybrevant is an intravenous (IV) formulation that requires dosing once every three weeks. Adding an IV formulation to the existing oral formulation, Leclaza, could double the effectiveness but reduce dosing convenience. Janssen is also developing a subcutaneous (SC) formulation to significantly reduce dosing time and address concerns about infusion-related side effects. Recently published clinical results showed that the combination of the subcutaneous formulation of Rybrevant SC+Leclaza achieved similar outcomes to Rybrevant IV+Leclaza. At a median follow-up period of 7 months, the Leclaza+Rybrevant SC was non-inferior to Leclaza+Rybrevant IV. “The Leclaza+Rybrevant combination therapy has continued to generate positive data in brain metastases, L858R mutation, etc., and therefore can be a viable first-line treatment option for EGFR-mutated NSCLC,” said a professor of medical oncology at a university hospital. ”It is difficult to say that use of the combination therapy is good for all patients, as the use of combination therapy is associated with more side effects than using either drug as monotherapy. This is why prescribing Rybrevantto to older patients can be challenging.” “The use of the intravenous formulation of Rybrevantrequires much attention. While a subcutaneous formulation is currently in development, the existing intravenous formulation is associated with frequent rashes, paronychia, and infusion-related adverse events. When selecting a treatment, we should consider not just the efficacy but the patient's overall experience, including side effects, frequency of visits, and quality of life. So there will be an active debate about which patients will benefit most from the early use of the combination therapy.” Concerns also rise on the lack of later-line therapies following the use of Tagrisso+platinum-based chemotherapy The use of Tagrisso and platinum-based chemotherapy as first-line treatment may lead to a shortage of treatment options for patients who develop resistance. In the past, EGFR-mutated NSCLC targeted therapies have been used as follows: 3rd generation TKIs were used on patients who were confirmed to be T790M-positive during biopsy after using 1st and 2nd generation TKIs, and platinum-based chemotherapy (Alimta plus carboplatin/cisplatin) was used on T790M-negative patients. So if a 3rd-generation TKI monotherapy is used in the first line, platinum-based chemotherapy is often used as a second-line treatment as they cannot use 1st or 2nd-generation TKIs due to resistance. This suggests that the use of both Tagrisso and platinum-based chemotherapy in the first line may lead to a lack of later-line treatment options. After developing a resistance to the combination, only taxane drugs such as docetaxel and paclitaxel and immuno-oncology drugs targeting PD-L1 will remain as treatment options. “After failing treatment with EGFR-TKIs, we usually use platinum-based chemotherapy, Alimta+cisplatin/carboplatin,” says a professor of medical oncology at a university hospital. “Combining these drugs with 3rd-generation TKIs is certainly therapeutic. However, we should also consider the lack of later-line treatment options after the patient develops TKI resistance if we pull the next line of treatment in advance amid a shortage of treatment options.” “As 3rd-generation TKIs have settled as the first-line standard of care, the role of 1st- and 2nd-generation TKIs will continue to diminish, and it is likely that it will ultimately become a competition between 3rd-generation TKI monotherapy and combination therapy.”
- Company
- Value of new drugs should be evaluated based on efficacy
- by Hwang, Byung-woo Aug 27, 2024 05:50am
- 'Receiving recognization of the value of new drugs' is one of the key challenges for pharma and biotech companies. With the proportion of new drugs being introduced into Korea rising and the number of homegrown new drugs increasing, the need for a drug pricing policy that recognizes the value of these new drugs has been growing. Recognizing the value of new drugs can lead to a virtuous cycle that drives profits and motivation for subsequent research and development (R&D) in the industry. The 'Measure to Improve the Drug Pricing System to Reflect the Innovative Value of New Drugs' being discussed in this context has raised industry expectations, promising improved access to innovative new drugs and preferential treatment during pharmacoeconomic evaluations. However, as the system is being implemented for the first time, a gray zone where the interpretation of the regulations may differ does exist, raising concerns about the system’s practicality. In the long run, it will be interesting to see if the government's intention to reflect the innovative value of drugs translates into the real world. 'Innovation' condition added to the flexible ICER threshold... Will the threshold exceed KRW 55 million? On the 16th, the National Health Insurance Review and Assessment Service (HIRA) released the 'Detailed Evaluation Criteria for Drugs Subject to Negotiation, including New Drugs', which was revised based on the deliberations of the Drug Reimbursement Evaluation Committee (DREC). The revisions include ▲ establishment of ICER threshold elasticity evaluation criteria for drugs ▲ addition of severe diseases to the risk-sharing program ▲ omission of the Drug Evaluation Committee when expanding the coverage of risk-sharing drugs under 1.5 billion won ▲ and new conditions for submitting clinical evidence such as RWD and RWE when renewing risk-sharing programs. The most notable part is that the 'innovativeness of the new drug' standard had been specified in the criteria to flexibly evaluate the ICER (incremental cost-effectiveness ratio) threshold. Previously, the regulations referred to the use of an ICER threshold as “instead of using an explicit threshold, flexibly refer to the results of previous deliberations considering the severity of disease, social burden of disease, impact on quality of life, and innovativeness.” The government’s In the specified regulations, a new drug is now regarded as ‘innovative’ when it satisfies all of the following criteria: ▲ there is no substitutable or therapeutically equivalent product or treatment ▲ a significant clinical improvement, such as prolonged survival, is recognized in the final outcome, ▲ the new drug is approved by the MFDS under Article 35(4)(2) of the Pharmaceutical Affairs Act through expedited review or is recognized as equivalent by the committee. The question now is how much of the innovativeness of a new drug can be recognized. The industry believes that Enhertu's case will serve as one reference point. Although there is no clearly documented figure, it is generally accepted that the maximum ICER granted for reimbursement in Korea is KRW 50 million. And even the KRW 50 million threshold is known to have been granted very few times. The ICER threshold that the government had set for Enhertu is likely in the low KRW 50 million range. In other words, the drug was recognized as cost-effective despite exceeding the ICER threshold. In this regard, the pharmaceutical industry believes that the ICER threshold should be more flexibly applied given the effectiveness of each drug. A representative from a multinational pharmaceutical company said, “If a drug has such a good effect that it prolongs survival, the ICER is bound to increase because it is more costly to administer. This should be recognized to some extent, but if the flexible ICER range is in the KRW 55 million range, these good drugs will still have difficulty passing the threshold.” For example, the ICER threshold should be more reflective of the innovativeness of a new drug that comes out after decades of lack of treatment, or a treatment that has a much better hazard ratio during survival than existing treatments and improves survival by over twofold. KRPIA’s research on the R&D Investment Status of Global Pharmaceutical Companies Actual cases of flexible ICER application is key...KRPIA, “Flexible approach adopted by major countries should be considered” In particular, the industry predicts that actual cases of application will be important, especially given that many drugs that should be eligible for flexible application of the ICER threshold are awaiting evaluation. “I think it's positive that the government explicitly stated their intention to recognize innovation compared to how such a wording was non-existent regarding the application of the ICER thresholds,” said an industry representative. “There are about 8 drugs that would want to be recognized for their innovativeness, so we have high hopes for the new direction.” “However, if the government’s range of flexibility is adding a mere KRW 5 million to the current upper ICER limit of KRW 50 million, a big difference in perspective will remain. The company's efforts are important, but for real-world application, it remains to be seen whether the government will be willing to recognize an ICER threshold of an unconventional level, whether it is KRW 70 million or KRW 100 million.” The industry pointed out that there are many difficulties for drugs in passing pharmacoeconomic evaluations, such as the reduction in the price of alternative drugs due to various follow-up measures and DREC’s repeated requests for supplementary data until the desired price level is reached. It is unlikely that a small increase in ICER, which fluctuates greatly depending on the assumptions made, will be enough to get an innovative new drug approved quickly. The ICER threshold desired by multinational pharmaceutical companies is reflected in the 'KRPIA Policy Proposal for Improving Public Healthcare' that was released by the Korean Research-based Pharmaceutical Industry Association (KRPIA). In the 'Prospects for New Drug Access Policy' part of the policy proposal, KRPIA suggested that it is not easy to objectively evaluate the effectiveness and innovativeness of new drugs and that it is necessary to consider the flexible new drug evaluation methods of major countries. KRPIA New Drug Access Policy Outlook (in comparison with major countries) In the United Kingdom, “a broad and multi-layered ICER criterion is applied, which considers the disease characteristics, clinical utility, payment value, and societal needs,” and in Canada “ICER is a reference for drug pricing negotiations, and the decisive factor is the level of symptom improvement, and the price of the drug is set at the upper limit of the median drug price in 11 major countries. In Germany, “pharmaceutical companies may autonomously set the initial price and negotiate reimbursement and discount rates after 1 year by evaluating its innovativeness and usage,” and Japan also adopts a system that evaluates a drug’s innovativeness and assigns an adjusted price. In consideration of this global trend, KRPIA emphasized that Korea also needs to more flexibly view ICER, etc. to recognize the innovativeness of new drugs. While the pharmaceutical industry welcomes the inclusion of the 'new drug’s innovative value' and 'expanded coverage for severe and rare diseases' in the government's major policies, the industry is emphasizing the need to set a more specific direction. For example, Kay Bae, the Chair of KRPIA stated, “While the government's recognition of the value of new drugs is an encouraging achievement, the government needs to come up with more practical and concrete measures so that new drugs can be supplied to patients quickly and lead to a virtuous R&D cycle in Korea.” The amended system has measures in place that require observation, such as the submission of clinical evidence such as RWD and RWE when renewing RSA contracts. In the long run, the government and the pharmaceutical industry are eye-to-eye in that they believe in the need to recognize the 'innovativeness of new drugs'. Well begun is half done, and the pharmaceutical industry welcomes the government’s launch of the 'Measure to Improve the Drug Pricing System to Reflect the Innovative Value of New Drugs.’ How well the system is applied and supplemented will remain a future task.
- Company
- K-similars won record number of U.S.·Europe nods this year
- by Chon, Seung-Hyun Aug 27, 2024 05:50am
- Korean biopharmaceutical companies have launched the highest number of biosimilars in history this year in the United States and European markets. As Celltrion and Samsung Bioepis won approvals for six biosimilars over eight months in the world's largest market, they topped the previous record of five cases of five years ago. According to sources on August 27th, Celltrion's SteQyma, a biosimilar to Stelara for the treatment of autoimmune diseases, received marketing authorization from the European Commission (EC). It reached the final commercialization step two months after the Committee for Medicinal Products for Human Use (CHMP) of the European Medicines Agency's (EMA) committee issued a positive opinion recommending SteQyma approval at the end of June. Stelara (ustekinumab), developed by Janssen, is a treatment for autoimmune diseases prescribed for plaque psoriasis, psoriatic arthritis, Crohn's disease, and ulcerative colitis. It inhibits the activity of interleukin (IL)-12,23, a type of inflammatory cytokine involved in immune responses. Last year, the global market size for ustekinumab was estimated at US$ 20.4 billion (about KRW 26.5 trillion). The European market was estimated at US$3.1 billion (about KRW 4.0 trillion), taking 15% of the global market. After Samsung Bioepis won European approval for Pyzchiva, a biosimilar to Stelara, in April, Celltrion entered the European market as the second Korean company. In June, Samsung Bioepis obtained a marketing authorization for Pyzchiva. U.S. and European biosimilar approvals obtained by Korean biopharmaceutical companies: (2024) Celltrion won European approval for Omlyclo, a biosimilar to Xolair, and SteQyma, a biosimilar to Stelara in 2024. Samsung Bioepis won European approval for Pyzchiva, a biosimilar to Stelara, and U.S. FDA approval for Opuviz, a biosimilar to Eylea, Pyzchiva, and Epysqli, a biosimilar to Soliris in 2024 (source: respective companies and FSS). Celltrion and Samsung Bioepis won six approvals for biosimilars in the United States and Europe this year. It exceeded the previous record of five approvals in 2019, breaking the highest number of approvals in history. Celltrion received a marketing authroziation for Omlyclo, a biosimilar to Xolair, from the EC in May. Omlyclo is the first Xolair biosimilar to receive the European marketing authorization. Xolair is an antibody biosimilar used to treat allergic asthma, chronic rhinosinusitis with nasal polyps (CRSwNP), and chronic spontaneous urticaria (CSU). Samsung Bioepis won four biosimilar approvals this year in the United States and Europe this year, with three approvals in the United States and one in Europe. In April, Samsung Bioepis won European approval for Pyzchiva, a biosimilar to Stelara. Since May, Samsung Bioepis succeeded in obtaining biosimilar approvals from the U.S. FDA for the past three months. It received approval for Opuviz, a biosimilar to Eylea. Eylea, developed by Regeneron in the United States, is indicated for treating wet age-related macular degeneration (AMD). Eylea generated approximately KRW 13 trillion in global sales last year. After winning FDA approval for Pyzchiva in June, Samsung Bioepis obtained a marketing authorization for its orphan drug Epysqli. Epysqli, developed by Alexion in the United States, is a biosimilar to Soliris. Epysqli won FDA approval as a treatment of paroxysmal nocturnal hemoglobinuria (PNH) and atypical hemolytic uremic syndrome (aHUS). In 2019, Celltrion and Samsung Bioepis received five biosimilar approvals in the United States and Europe. In November 2019, Celltrion received European approval for Remsima SC, a subcutaneous injection formulation of Remicade. Remsima SC is a biosimilar developed by Celltrion by changing the formulation of Remicade from intravenous (IV) to subcutaneous (SC) injection. In 2019, Samsung Bioepis received FDA approval for four biosimilars to Herceptin, Enbrel, Humira, and Lucentis. In January 2019, Ontruzant, a biosimilar to Herceptin, received a marketing authorization in the United States, followed by Eticovo and Hadlima, biosimilars to Enbrel and Humira, in April and July of the same year, respectively. Samsung Bioepis obtained FDA approval for its Byooviz, a biosimilar to Lucentis, in the United States in September 2021. Korean pharmaceutical companies have started actively targeting the global biosimilar market after Celltrion's Remsima received European marketing authorization under the title of 'The world's first antibody biosimilar' in August 2013. Since 2016, Korean biopharmaceutical companies continued to win new approvals yearly in the United States and Europe. Celltrion achieved success, obtaining eight approvals in Europe and six in the United States. The company also entered the European market for Remicade, Mabthera, Herceptin, Avastin, Humira, Xolair, and Stelara. In 2016, Celltrion's Inflectra, a biosimilar to Remicade, became the first to win FDA approval in the United States. In 2018, Truxima and Herzuma received FDA approvals. In September 2022, Celltrion obtained a marketing authorization for Vegzelma, a biosimilar to Avastin, from the FDA, and last year, Yuflyma, a biosimilar to Humira, also received FDA approval. In August last year, Celltrion received a marketing authorization for Zymfentra, a SC formulation of Remsima, as a novel drug. Samsung Bioepis won eight approvals in Europe and six in the United States. In January 2016, Samsung Bioepis began its global market expansion by obtaining approval for its Benepali, a biosimilar to the autoimmune disease treatment Enbrel, in Europe. Afterward, the company received European approval for biosimilars referencing Remicade, Herceptin, Humira, Avastin, Lucentis, Soliris, and Stelara. In April 2017, Samsung Bioepis received the first FDA approval in the United States for its Renflexis, a biosimilar to Remicade. After that, the company entered the U.S. market for biosimilars referencing Herceptin, Enbrel, Humira, Lucentis, Eyela, Stelara, and Soliris.
- Policy
- SK Chemicals strengthens migraine drug lineup with Suvexx
- by Lee, Tak-Sun Aug 27, 2024 05:50am
- Suvexx, a combination migraine treatment imported and supplied by SK Chemicals, will be reimbursed in Korea next month. Suvexx is a combination of sumatriptan, a triptan-class drug most commonly used for migraine, and naproxen, a non-steroidal anti-inflammatory drug (NSAID). According to industry sources on the 26th, Suvexx will be listed for reimbursement at KRW 5,042 per tablet starting on the first of next month. There has never been a combination drug combining sumatriptan naproxen in Korea. Furthermore, the individual ingredients, sumatriptan succinate 85mg and naproxen sodium 500mg, are not registered on the reimbursement list. Therefore, HIRA applied a formula based on the upper insurance price limit of products that contain similar dosages of the ingredients to calculate the upper limits of each component and then added them up. The resulting price was KRW 5,042 per tablet. The total price is higher than the maximum price of KRW 3,615 set for sumatriptan succinate 50 mg and KRW 420 for naproxen 1 g, which are currently registered on the reimbursement list. Suvexx is indicated for the treatment of acute migraine attacks with or without prodromal symptoms in adult patients 18 years of age and older. In clinical trials, a significantly higher proportion of subjects achieved headache relief 2 hours after dosing compared to placebo, and the proportion of patients who remained pain-free for 24 hours after dosing without taking any other medication was significantly higher in the treatment group compared to placebo, sumatriptan monotherapy, and naproxen monotherapy, demonstrating the drug’s efficacy. The product was co-developed by global pharmaceutical giant GSK and Pozen, a subsidiary of Canada's Aralez Pharmaceuticals, and SK Chemicals signed a contract with the original developer to introduce the drug in Korea in 2021. SK Chemicals received domestic approval for the drug on August 1 last year. SK Chemicals has been supplying various products in the domestic migraine market. Recently, it co-marketed Emgality, a migraine prevention injection that targets calcitonin-gene-related peptide (CGRP), with Lilly. The company also owns the triptan-based Migard Tab (frovatriptan), so the launch of Suvexx is expected to create synergy, strengthening the company’s product lineup. The annual prescription volume of triptan-based migraine drugs is approximately KRW 20 billion.

- InterView
- AstraZeneca seeks mutual growth through open collaboration
- by Hwang, Byung-woo Aug 27, 2024 05:50am
- Multinational pharmaceutical companies are increasingly investing in R&D in Korea’s domestic pharmaceutical industry through open innovation. According to the Korean Research-based Pharmaceutical Industry Association (KRPIA), the total amount of R&D invested in clinical research in Korea in 2022 was KRW 817.8 billion and has been on the rise for 3 consecutive years. AstraZeneca Korea, which recently introduced bold new drugs such as Enhertu and Imfinzi, is also aiming to create a virtuous cycle of shared growth with the domestic pharmaceutical industry through active R&D investment. AZ invests more than 30% of sales in R&D...makes notable achievements in developing innovative drugs AstraZeneca Korea's sales surpassed the KRW 600 billion mark in 2021 (KRW 655.3 billion), based on audited financial statements. At the time, sales were boosted by its COVID-19 vaccine, but the company's robust anti-cancer portfolio and rare disease therapies acquired through the acquisition of Alexion have since become new growth drivers, posting sales of KRW 615.1 billion in 2022 and KRW 639.3 billion in 2023. AstraZeneca Korea's sales growth is significant because the company has invested more than 30% of its revenue back into the domestic industry. According to the company, it invested KRW 215 billion in Korea in 2023, which is about 34% of its annual revenue. Of this, KRW 116 billion was spent on clinical research. This coincides with the discussions it had made at the Korea-Sweden Business Summit that was held in Stockholm, Sweden in 2019. At that time, AstraZeneca announced a plan to invest KRW 850 billion in Korea, which it implemented for 5 years. AstraZeneca Korea AstraZeneca's investment in Korea is particularly noteworthy because it is not limited to R&D. For example, the global clinical TOPAZ-1 trial of the immuno-oncology drug Imfinzi, which was led by Dr. Do-Yoon Oh, professor of Medical Oncology at Seoul National University Hospital, changed the global biliary tract cancer treatment paradigm by showing the potential to improve survival outcomes in biliary tract cancer, a disease with an average survival period of less than one year with existing treatments. In 2023, the company co-developed and launched Sidapvia, a combination diabetes drug, after 4 years of collaboration with SK chemicals, and is currently working together for its global commercialization. AstraZeneca Korea is also expanding its collaboration with the Korean government to strengthen domestic research capabilities. The company has been running the ‘KHIDI-AZ Anti-Cancer Research Support Program’ for over a decade, a program that selects and supports research projects in the field of anti-cancer with the Korea Health Industry Development Institute (KHIDI), with the goal of overcoming cancer, the No.1 cause of death in Korea. AstraZeneca Korea Four of the projects that have been completed under the program have led to tangible results, including the publication of▲SCI papers, ▲lectures at the official international conferences of the Korean Diabetes Association and the Asian Association for the Study of Diabetes, and ▲publications in the Journal of the Korean Diabetes Association. As a result of these open innovation achievements, the company has been selected as an 'innovative pharmaceutical company accredited by the Ministry of Health and Welfare' for 6 consecutive years since 2018. As of June, there are only 3 multinational pharmaceutical companies that fall in the innovative pharmaceutical company category as announced by the Ministry of Health and Welfare. Aims to create a patient-centered care ecosystem and rebuild community for development Ultimately, AstraZeneca Korea believes that beyond the development and supply of innovative medicines, it is necessary to address the blind spots in treatment that occur in the pre-approval and reimbursement stages. Examples include attracting clinical trials to Korea so that patients can start treatment with innovative medicines that are not yet available in the country, and running patient support programs to increase patient access to medicines that are yet to be reimbursed in Korea. As of 2023, AstraZeneca Korea is conducting approximately 130 clinical trials in Korea, making it the pharmaceutical company with the most clinical trials approved by the Ministry of Food and Drug Safety (MFDS), excluding CROs. Through AstraZeneca's clinical trials, about 2,600 cancer patients in Korea have received new anti-cancer drugs in the last 5 years (2018-2023), and about 1,004 patients with ultra-rare diseases have been treated through a total of 37 clinical trials in the last 6 years (2019-2024). The company also continuously expands support to minimize treatment gaps. Currently, more than 10 early access programs are in place, and it is known that about 260 cancer and extremely rare disease patients have received treatment through these programs so far. Examples of AstraZeneca Korea “AstraZeneca is committed to the development and supply of innovative medicines through increased investment in Korea, including in R&D talent,” said Sewhan Chon, Country President of AstraZeneca Korea. ‘We are constantly striving to create a healthcare ecosystem that can mutually grow with our community across the pharmaceutical industry, medical research, and patients’ lives.”
- Company
- More urothelial cancer drug options now available in KOR
- by Hwang, Byung-woo Aug 26, 2024 05:46am
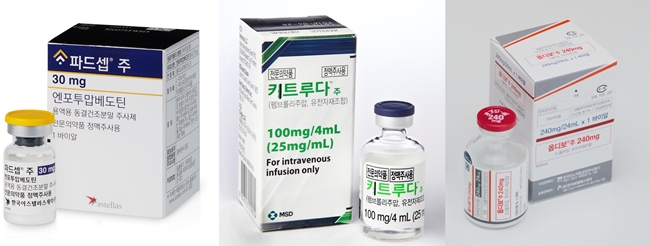
- A number of immuno-oncology and antibody-drug conjugate (ADC) drugs are being granted approval as a first-line treatment of urothelial cancer, increasing the treatment options that had previously been dominated by platinum-based chemotherapy. Although they are not yet reimbursed and have yet to become mainstream treatment options, it is expected that the strategy for each line of treatment will change in the long term. (from left to right) Pic of Padcev, Keytruda, Opdivo Urothelial carcinoma starts in the epithelial cells lining the urinary tract and is the most common type of bladder cancer, accounting for 90% of all bladder cancer diagnoses. However, unlike other cancers like lung and breast cancer, where the standard of care changes quickly with the introduction of new drugs, UC has remained a barren area for decades, leaving a large unmet need for first-line treatment options. For the past 30 years, platinum-based chemotherapy has remained the first-line standard of care for UC. The introduction of Opdivo (nivolumab) has changed the situation. The drug was approved by the Ministry of Food and Drug Safety on the 17th as a first-line treatment for unresectable or metastatic urothelial carcinoma in combination with cisplatin and gemcitabine (GemCis). The approval was based on results from the Phase III CheckMate 901 trial in patients with unresectable or metastatic UC who had not received prior therapy. Results showed that at a median follow-up of 33.6 months, the primary endpoint, median OS (mOS), was 21.7 months with the use of the Opdivo+GemCis combination, which is significantly longer than the 18.9 months in the GemCis only arm, and reduced the risk of death by 22%. In particular, the CheckMate-901 pivotal trial, which is evaluating Opdivo in combination with Yervoy (ipilimumab) versus the standard of care, leaves room for further expansion of Opdivo’s indications. Keytruda (pembrolizumab) + Padcev (enfortumab vedotin), which received approval from the MFDS on March 2 5 following Opdivo’s approval, is also expected to rise as a first-line treatment option in UC. Highlighted as a novel combination of an immuno-oncology drug and an ADC, the Keytruda-Padcev combination gained approval as the first treatment to change the first-line treatment paradigm for UC in 30 years when the first data were presented at the European Society for Medical Oncology Annual Meeting (ESMO 2023) last year. The Phase III EV-302/KEYNOTE-A39 trial, which became the basis of its approval, the combination demonstrated a median progression-free survival (PFS) of 12.5 months over a median follow-up of 17.2 months, with a 55% reduction in the risk of disease progression and death compared to 6.3 months in the placebo arm. Dr. Jae-Lyun Lee, Professor of Medical Oncology at Seoul Asan Medical Center, said, “No other treatment in UC has shown this level of efficacy in the past 30 years. The more than twofold increase in progression-free survival is truly remarkable, which is why we have high hopes for the Keytruda-Padcev combination. Based on the clinical trial results, Padcev+Keytruda is expected to rise to the forefront among first-line treatment options on-site. Currently, Bavencio (avelumab) is reimbursed as a first-line maintenance therapy, but as new first-line treatment options become available, a change in the treatment regimen and later-line therapies is inevitable. However, Opdivo, which was added to existing first-line treatment options, and Keytruda+Padcev, an immuno-oncology drug, and an ADC combination, both are yet to be reimbursed in Korea, leaving the high cost an issue. Dr. Inho Kim, professor of Medical Oncology at St. Mary's Hospital in Seoul, said, “Drugs like Padcev have shown good results recently, and I think each drug has its pros and cons. Cost is also a consideration, and as we gain more prescription experience and accumulate more information on the patients’ conditions, we will be able to set guidelines for this.”
- Company
- ‘Lilly will reapply for Retevmo’s reimbursement in Korea’
- by Eo, Yun-Ho Aug 26, 2024 05:46am
- “We will do our best to receive reimbursement for Retevmo in Korea. However, the will of the health authorities is as important” The company has expressed its will to list a RET-targeted therapy option in Korea. The question that remains now is when. Lilly's RET inhibitor Retevmo (selpercatinib) remains non-reimbursed since the company’s pricing negotiations with the National Health Insurance Service broke down last year There are only two RET-targeted therapies - ‘Gavreto (pralsetinib),’ which Roche Korea introduced from Blueprint Medicines, and ‘Retevmo (selpercatinib)’ by Lilly Korea – currently approved in Korea. Of the two, it would be hard for Gavreto to be listed for reimbursement in Korea as Roche gave up rights to the drug. The situation is not so better off for Retevmo either. Retevmo, which was approved in Korea in March 2022, failed to pass the Health Insurance Review and Assessment Service Cancer Disease Review Committee (CDDC) review in May of the same year, but then passed CDDC review in November and finally passed the Drug Reimbursement Evaluation Committee in May last year. After passing the DREC review, the company entered into drug price negotiations with the National Health Insurance Service in June, raising expectations on Retevmo’s reimbursement. However, the two ultimately failed to reach an agreement. In fact, it was the only news of a drug pricing negotiation failure reported in the past year. Retevmo was granted marketing authorization under the condition of conducting a Phase III trial, so it had trouble during the reimbursement listing process as the authorities requested the company for data equivalent to a Phase III trial to accommodate for the lack of its Phase III trial data. At the time, this led to criticism about the reimbursement evaluation criteria for conditionally approved drugs that were granted fast-track review. The reimbursement of Retevmo, which applied for fast-track status under the approval-reimbursement evaluation linkage system, had been in discussions for about a year and a half, but to no avail. But it now has additional results from a proper Phase III trial. Now it's a matter of Lilly and the government’s willingness to bring RET drugs to the market. Results from Phase III trials on Retevmo - LIBRETTO-431 and LIBRETTO-531 - were presented at the European Society for Medical Oncology Annual Congress (ESMO 2023) last year. The results were published in the internationally recognized New England Journal of Medicine (NEJM) along with the congress presentation. The LIBRETTO-431 trial presented at the meeting compared Retevmo with platinum-based chemotherapy±pembrolizumab as a first-line treatment in patients with advanced or metastatic RET fusion-positive NSCLC. Key findings in the trial showed that in the ITT-pembrolizumab population, the median progression-free survival (PFS) by an independent centralized review committee (BICR) was 24.8 months in the Retevmo arm, and 11.2 months in the control arm, with a hazard ratio of 0.465. The overall response rate (ORR) by BICR was 83.7% in the Retevmo arm, which was statistically significantly higher than the 65.1% in the control arm. “Lilly intends to reapply for reimbursement of Retevmo,” said a Lilly representative. However, for us to quickly reapply for reimbursement, not only the company's efforts but also discussions with health authorities are required. So we cannot provide a specific timeline for the reapplication yet. We remain committed to improving access to Retevmo for cancer patients with RET gene mutations in Korea.” In 2020, Retevmo was approved as the first treatment option for cancer patients with RET alternations in the US after the FDA reviewed the drug through the Accelerated Approval and Priority Review pathway and granted the Breakthrough Therapy & Orphan Drug Designation.
- Policy
- Eliquis generics re-enter the market, 35 reimbursed drugs
- by Lee, Tak-Sun Aug 26, 2024 05:46am
- Product photo of the original drug Eliquis. Drugs that are generic versions of the coagulant agent Eliquis (apixaban) will re-enter the market three and five months after discontinuing sales due to a failing patent nullification challenge. It is because the original drug's substance patent is set to expire on September 9th. According to sources on August 23rd, generics containing apixaban will be listed for reimbursement on September 10th. 18 drugs from 35 pharmaceutical companies will be reimbursed. The following companies will be launching their products in the market: Kyongbo Pharmaceutical, Medica Korea, Boryung, Vivozon Pharmaceutical, Ilhwa, Chong Kun Dang Pharmaceutical, Huvist Pharmaceutical, Huons, Daewoong Bio, Dong Kwang Pharmaceutical, Dongkook Pharmaceutical, Samjin Pharmaceutical, Shinil Pharma, Alicon Pharmaceutical, Genuone Sciences, Hana Pharm, Hutecs Korea Pharmaceutical, and Hanlim Pharmaceutical. Eliquis generics have been on the market since June 2019 after the Supreme Court decision. Generics won the substance patent nullification trial and the Supreme Court trial. However, the situation reversed. In April 2021, the Supreme Court ruled against the previous decision and destroyed the case in favor of the original company. Consequently, products that could potentially infringe on the patent were withdrawn from the market in two years. The original Eliquis' price was restored. Chong Kun Dang Pharmaceutical's Liquisia recorded KRW 4.1 billion (based on UBIST) in outpatient sales during the sales. Chong Kun Dang Pharmaceutical's Liquisia aims to regain the market presence. The price of drugs meeting the requirement criteria and receiving 59.5% credit as first generics are set as KRW 633 per tablet. Drugs meeting only one requirement criteria are set as KRW 484, which is 45.52% of the highest price. Boryung's Viala Fix will be listed as KRW 724, a 68%, credited as the innovative drug. Products that are set for listing have been previously launched. Some are new products. A pharmaceutical industry official said, "Eliquis was worth KRW 50 billion when listed in 2020. After generics withdrew from the market due to the Supreme Court ruling in 2021, Eliquis' price was restored. Therefore, it became the blockbuster drug with KRW 77.3 billion in prescription sales last year, based on UBIST," and added, "The market is expected to grow when new anticoagulants become prescribed in private practices. As a result, Korean pharmaceutical companies will actively pursue sales and marketing."
- Company
- Colorectal cancer drug Fruzaqla may be introduced in H2
- by Eo, Yun-Ho Aug 26, 2024 05:46am
- A new treatment option is expected to emerge in the field of colorectal cancer later this year. According to industry sources, the final review is underway for the approval of Fruzaqla (fruquintinib), which was designated as a Global Innovative products on Fast Track (GIFT) by the Ministry of Food and Drug Safety in November last year. Fruzaqla was designated as an orphan drug in Korea in February and was granted priority review by the U.S. FDA in May last year and received final approval in November of the same year. In addition to the U.S. FDA approval in November last year, the drug recently received final approval from the European Commission. Fruzaqla is specifically indicated for the treatment of adult patients with metastatic colorectal cancer (mCRC) who have been previously treated with fluoropyrimidine-, oxaliplatin-, and irinotecan-based chemotherapy, an anti-VEGF treatment, and if RAS wild-type, an anti-EFGR treatment (RAS); and at least one of trifluridine plus tipiracil or regorafenib treatment. Fruzaqla (fruquintinib) is a VEGFR-1, -2, -3 receptors inhibitor that Takeda Pharmaceutical acquired the rights to from Hong Kong Hutchmed. Fruzaqla’s approval was supported by the FRESCO trial, which was conducted in China and published in JAMA, and the global FRESCO-2 trial that was published in LANCET. Both studies compared Fruzaqla combination therapy (best supportive care (BSC)) to placebo combination therapy in previously treated mCRC patients. The results showed that both FRESCO and FRESCO-2 studies met their primary and important secondary endpoints and showed consistent benefits in a total of 734 patients treated with Fruzaqla. In the FRESCRO-2 clinical trial, the fruquintinib-treatment group yielded a median overall survival (OS) of 7.4 months, versus 4.8 months for the placebo group. In the FRESCO clinical trial, the fruquintinib-treatment group yielded a median OS of 9.3 months, versus 6.6 months in the placebo group. The MFDS grants the GIFT designation to ▲ drugs aimed at treating serious life-threatening diseases such as cancer or rare diseases ▲ drugs aimed at preventing or treating infectious diseases that may cause serious harm to public health, such as bioterrorism infectious diseases or pandemics, ▲ new drugs developed by Korea Innovative Pharmaceutical Companies designated by the Ministry of Health and Welfare, ▲ drugs used in combination with medical devices subject to expedited review. Drugs subject to GIFT can receive various support that accelerates commercialization, including: ▲support for regulatory approval, ▲rolling review, ▲close communication between the reviewer and developer through product presentation and supplementary briefing sessions, and ▲expert consulting by regulation experts.
- Policy
- Hanmi faces competition from cheaper Zytiga generic drug
- by Lee, Tak-Sun Aug 26, 2024 05:45am
- Product photos of Janssen A new competing drug has entered the 'Zytiga' generic market, where Hanmi Pharmaceutical was the only company to launch a product last year. Ace Pharmaceutical has introduced a drug imported from India to the South Korean market. While Hanmi Pharmaceutical is strengthening its market presence by recently launching a combination drug, the new generic entry garners attention due to its impact on market competition. According to industry sources on August 25th, Ace Pharmaceutical listed 'Aviron Tab 500 mg (abiraterone acetate)' for reimbursement. The drug has the same active ingredient as Janssen Korea's Zytiga. Zytiga is an anticancer used in treating three types of prostate cancer: ▲patients with asymptomatic or mildly symptomatic metastatic castration-resistant prostate cancer (mCRPC), ▲patients with mCRPC previously treated with docetaxel, ▲patients newly diagnosed with hormone-sensitive metastatic prostate cancer (mHSPC) in combination with androgen deprivation therapy (ADT) plus prednisolone. The drug is expected to be used more as the patient burden decreases with the transition from selective reimbursement coverage (30% co-payment rate) to essential reimbursement coverage (5% co-payment rate). It generated KRW 19 billion last year, down -13%p year over year (YoY), based on IQVIA. Such a decrease in Zytiga's sales may be due to the generic entry. Hanmi Pharmaceutical launched 'Abiteron Tab 500 mg,' the first Zytiga generic in South Korea, last October. Last month, Hanmi Pharmaceutical launched a combination drug, 'Abiterone Duo Tab,' a combination of Zytiga and prednisolone. The company developed the product after noting that reimbursable Zytiga therapy can be combined with prednisolone. The industry draws attention to whether Hanmi Pharmaceutical can establish a presence in the market for anticancer agents with its proprietary combination drug. However, the company encountered a surprise. Ace Pharmaceutical, which imports foreign anticancer agents to South Korea, has recently introduced Zytiga generic. Ace Pharmaceutical has recently distributed 'Actepa Inj (thiotepa),' used as a conditioning regimen for allogeneic or autologous stem cell transplantation before the treatment of adult lymphoma and pediatric neuroblastoma or retinoblastoma, to Asan Medical Center. Therefore, the company demonstrated competitiveness in the market for anticancer agents. Aviron Tab's drug price is less expensive than 'Abiteron.' It was listed for reimbursement as KRW 8,498 per tab, which is lower than the calculated drug price. Aviron Tab's drug price is lower than KRW 8,537 for Abiteron tab and substantially differs from the original Zytiga tab (KRW 11,746). While original drugs show a strong presence in the market for anticancer agents, the competition in the market sized KRW 20 billion is expected to heat up after the entries of the first generic, Hanmi Pharmaceutical's combination drug, and a new generic.