- LOGIN
- MemberShip
- 2026-06-20 14:31:20

- Product
- Generic substitutions and INN-based prescriptions on track
- by Kim JiEun Sep 04, 2025 06:12am
- The prolonged instability in drug supply has fueled momentum in the pharmacy community, with both streamlined generic substitutions and limited mandatory international nonproprietary name prescriptions gaining traction in the National Assembly. On September 2, Democratic Party of Korea lawmaker Jong-Tae Jang introduced amendments to the Medical Service Act and the Pharmaceutical Affairs Act. The proposed bill would create a legal definition for supply-unstable medicines and mandate ingredient name-based prescriptions for such drugs on a limited basis. This follows the passage of the so-called “Generic Substitution Activation Act,” recently approved by the National Assembly’s Health and Welfare Committee, adding yet another long-pursued initiative of the pharmacy sector to the legislative agenda. The shift began with revisions to the enforcement regulations of the Pharmaceutical Affairs Act, which simplified the notification process for generic substitutions. Despite strong opposition from physicians, the Ministry of Health and Welfare pushed through the revision, allowing the HIRA (Health Insurance Review and Assessment Service) online portal as one of the means for post-notifications. Even with doctors voicing concerns of “infringement of prescribing rights,” the government and the National Assembly have continued to push for regulatory revisions and legislation due to the persistent issue of drug shortages. The issue, which first emerged during the early COVID-19 period, have remained unresolved even 5 years after the pandemic subsided. Critics argue the government has failed to present a clear solution. As a result, consensus between the government and the National Assembly on the need to address drug shortages has directly driven attempts at systemic reform. During the last presidential election, the Democratic Party of Korea pledged to implement limited INN-based prescriptions for essential medicines as part of its project to secure a stable drug supply. This confirmed the Lee Jae Myung administration’s commitment to addressing the shortage issue. In its May policy platform, the Democratic Party of Korea included “promoting generic substitutions and limited ingredient-name prescriptions for essential drugs with unstable supply” under its measures to ensure stable access to essential medicines. The pharmacist community believes the streamlining generic substitution law will pass smoothly, given that regulatory changes have already been enacted. However, they remain cautious on mandatory INN-based prescriptions, anticipating fierce resistance from physicians. The Korean Medical Association is likely to intensify its opposition and pressure the government. A pharmacy association representative commented, “We welcome the fact that bills addressing drug shortages are being introduced and advanced with a sense of urgency. For years, responses to shortages have been nothing more than post-hoc measures. Now is the time for preventive and systemic solutions. Although past attempts at generic substitution and INN-based prescriptions repeatedly collapsed due to opposition from the medical community, we are hopeful that with shortages now recognized as a societal issue, this time will be different.”
- Policy
- Pilot Drug Approval·Review Coordination Committee extended
- by Lee, Hye-Kyung Sep 04, 2025 06:10am
- The pilot operation period for the Pharmaceutical Approval and Review Coordination Committee will be extended by one year. According to the pharmaceutical industry on September 3, the Ministry of Food and Drug Safety (MFDS) decided to extend the pilot period for the Coordination Committee by one year, starting from August 29. The committee had been operating on a trial basis for one year since June 17 of the previous year. This extension is based on a revision of the committee's operational guidelines. For the past year, applications for adjustment were limited to cases involving safety and efficacy review data, quality review data, and documentation related to data protection. However, starting this year, the scope has been expanded to include 'all matters' for which the Pharmaceutical Approval Management Division has requested supplementary data. The scope of applications for adjustment following the Pharmaceutical Approval and Review Coordination Committee extension. The Coordination Committee was established last June to make the drug product approval and review process more transparent and rational. It allows applicants to request an adjustment when supplementary data is asked for during the drug product approval and review process. The committee, chaired by the Director of the Drug Safety Bureau, includes the Head of the Pharmaceutical Review Department, the Director of the Pharmaceutical Policy Division, the Director of the Pharmaceutical Approval Management Division, relevant review department heads, and experts from the Central Pharmaceutical Affairs Council. They proceed with adjustments through a majority vote. With the pilot program extended for another year, it will apply to applications submitted to the Pharmaceutical Approval Management Division. The MFDS plans to evaluate the operational results at the end of this period to decide whether to terminate the pilot program, transition it to a full-scale operation, or further expand the scope of adjustment applications. The scope of applications for adjustment includes ▲Review data specified in Article 5 of the 'Regulations on Drug Product Approval, Notification, and Review' ▲Data related to the eligibility for clinical trial data protection, as per Article 31-6 of the 'Pharmaceutical Affairs Act' and Article 21-2 of the 'Rules on the Safety of Pharmaceuticals, etc.' ▲Other matters for which adjustment is deemed necessary in response to a request for supplementary information (excluding requests for supplementary data needed for evaluating the implementation status of Good Manufacturing Practice (GMP) and clinical trials). An applicant wishing to request an adjustment must submit an application form, as specified in Annex No. 1 of the guidelines, to the Pharmaceutical Approval Management Division within 30 days of the request for supplementary information. The committee plans to process the adjustment applications, in principle, within the 60-day supplementary request period. Only the applicant listed on the application form can request an adjustment; an agent cannot make this request. Meanwhile, once an application is selected for adjustment, it will be thoroughly discussed at a Coordination Committee meeting, which is composed of both internal and external experts. A two-thirds majority vote of the committee members present decides on the supplementary requirements. The results of the adjustment are then communicated to the relevant divisions and the applicant.
- Policy
- MFDS allocates KRW 812.2 billion for next year's budget plan
- by Lee, Hye-Kyung Sep 04, 2025 06:09am
- The Ministry of Food and Drug Safety (MFDS; Minister, Yu-Kyoung Oh) has announced that its budget for 2026 has been set at a total of KRW 812.2 billion, an increase of KRW 63.3 billion (8.4%) from this year's budget of KRW 748.9 billion. This budget plan was focused on facilitating the smooth execution of the new government's national tasks and restructuring expenditure to ensure efficient financial management. The 2026 MFDS budget plan includes four areas: enhancing safety and expanding the foundation for innovative growth in the pharmaceutical and bio-health sectors, strengthening customized safety support for food and medicine, considering the regulatory environment, creating a safe food and healthy dietary environment, and building a proactive food and drug safety management system for the future. A total of KRW 170.4 billion has been allocated to enhance safety and expand the foundation for innovative growth in the pharmaceutical and biotech sectors. The Fund for the Korea Orphan & Essential Drug Center will increase from KRW 4.5 billion this year to KRW 6.7 billion next year. To resolve the unstable supply of rare and essential medicines, MFDS will strengthen the stable supply foundation by expanding contract manufacturing of discontinued products and the emergency import of self-administered medicines with minimal demand. For supporting and building a management system, such as innovative medical devices, KRW 2 billion will be invested. A newly allocated budget of KRW 15 billion will be invested next year to fund the rapid commercialization of AI-driven products. MFDS will assist in the swift commercialization of promising AI-based products in the food and medical device sectors, thereby shortening Korean companies' development timelines and accelerating their market entry. The budget for the Korean Association Against Drug Abuse has been increased from KRW 16.5 billion this year to KRW 17.1 billion next year. A total of KRW 105.4 billion has been allocated for strengthening customized safety support for food and medicine, taking into account the regulatory environment. With the biohealth industry continuing to grow and the demand for systematic regulatory support from industries with limited experience and expertise increasing, MFDS plans to expand regulatory support by building an integrated consultation platform and securing customized consultation personnel for advanced and next-generation biopharmaceuticals. The budget for this will be significantly increased from KRW 500 million this year to KRW 11.4 billion next year. To lead the development of the pharmaceutical industry, MFDS will create review guidelines for new technologies and concepts, such as AI-driven products, and establish review standards that consider the characteristics of advanced and next-generation biopharmaceuticals. This will secure approval and review capabilities at the level of developed countries. To counter non-tariff barriers, such as country-specific approval regulations that hinder the export of Korean pharmaceuticals, MFDS will operate export approval support hubs. These hubs will analyze and provide case studies of approvals by product and offer regulatory consultations for export countries, thereby supporting the swift acquisition of overseas drug approvals. A new budget of KRW 5.5 billion has been allocated for operating a regulatory science talent development program in which universities, industries, and research institutes participate. The goal is to cultivate regulatory science professionals who can scientifically evaluate the safety of advanced bio-health products. A total of KRW 187.1 billion has been allocated for creating a safe food and healthy dietary environment, and a total of KRW 146.9 billion has been allocated for building a food and drug safety management system for the future. Next year, MFDS plans to build an automated drug approval and review system. This system will verify submitted document requirements, handle repetitive and routine tasks, and generate data summaries and reports. The system is intended to address the shortage of review personnel and expand patients' treatment opportunities through a faster drug approval process. The automation system will be expanded from generic drugs next year to active pharmaceutical ingredients (APIs) in 2027 and new drugs in 2028. To respond to the changing environment, such as online food distribution and the development of artificial intelligence, MFDS will establish an Information Strategy Plan (ISP) to build an "Integrated Food Safety Information Network." This network will consolidate 15 food-related information systems and automate public service and administrative tasks. In addition, as the scope of narcotics investigations expands, MFDS will secure digital forensics personnel and equipment dedicated to investigating medicinal narcotics. It will also broaden the synthesis of standard materials for new psychoactive substances and the evaluation of dependence on temporary narcotics. MFDS stated, "Once the 2026 budget plan is finalized through the National Assembly's deliberation process, we will do our best to execute the new government's national tasks and key projects without delay and protect the health and safety of the public."
- Company
- Novo Nordisk, Kakao Health sign digital healthcare MOU
- by Cha, Jihyun Sep 04, 2025 06:09am
- Kakao Healthcare (CEO Hee Hwang) announced on the 3rd that it has signed a memorandum of understanding with the multinational pharmaceutical company, Novo Nordisk Korea (General Manager Kasper Roseeuw Poulsen) to build a digital healthcare ecosystem for obesity and diabetes patients. The signing ceremony, held on September 2 at Novo Nordisk Korea’s headquarters in Songpa-gu, Seoul, was attended by Kakao Healthcare CEO Hee Hwang and Novo Nordisk Korea CEO Kasper Rosseeu Poulsen. At the event, the two companies agreed to collaborate by utilizing various digital technologies to improve the treatment journey of obesity and diabetes patients, ultimately creating a sustainable treatment environment that can deliver better outcomes. This is the second collaboration between the two companies, following their first partnership in 2023. Under the agreement, they linked Kakao Healthcare’s AI-based health management app ‘Pasta’ with Novo Nordisk’s insulin FlexTouch pen and its smart cap ‘Mallya,’ providing a medication management solution for diabetes patients. The new partnership is significant in that it expands the scope of collaboration from diabetes to obesity, creating a comprehensive alliance. The companies plan to address the unmet medical needs of Korea’s rapidly growing obesity and diabetes populations by offering patient-centered digital solutions. Specifically, in the field of obesity, the partnership will focus on ▲developing a personalized digital support program, ▲providing solutions to improve treatment effectiveness and quality of life. and In particular, the partnership will integrate Novo Nordisk’s patient support program Novo Fit Care, which is offered to patients prescribed its obesity treatments, into the Pasta app. Through this, patients will be able to manage weight and other long-term health aspects in a comprehensive way. In the field of diabetes, the companies will jointly develop digital solutions aimed at improving disease awareness. Kasper Roseeuw Poulsen, General Manager of Novo Nordisk Korea, said, "Obesity and diabetes are chronic conditions which, if left untreated, can lead to serious comorbidities and impose a tremendous burden on individuals and society. With Novo Nordisk’s more than 100 years of dedication to diabetes and obesity, and Kakao Healthcare’s leadership in Korea’s digital healthcare sector, the partnership will further accelerate integrated innovation for patient support in Korea." Hee Hwang, CEO of Kakao Healthcare, said, "Kakao Healthcare has leveraged AI, big data, and other technologies to bring positive changes to patients’ health journeys. By combining this experience and technology with Novo Nordisk, a global leader in obesity and diabetes care, we aim to play a major role in expanding patient-centered digital healthcare on a global scale."
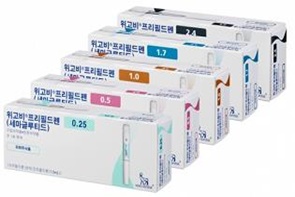
- Policy
- HIRA "Reimb application of Wegovy has not been filed"
- by Lee, Jeong-Hwan Sep 04, 2025 06:08am
- Product photo of Wegovy The Health Insurance Review & Assessment Service (HIRA) announced that it will conduct a fair and swift evaluation if an application for reimbursement is submitted for Novo Nordisk's popular obesity drug, Wegovy (semaglutide). HIRA clarified that since Wegovy's company has not yet applied for reimbursement, HIRA is yet at the stage of determining Wegovy's National Health Insurance reimbursement. HIRA stated recently after a recent inquiry from Rep. Kim Seon-min of the Cho Kuk Innovation Party regarding the reimbursement status of Wegovy. Rep. Kim had asked HIRA for its stance on a plan to transition the non-reimbursed prescription drug Wegovy to National Health Insurance coverage and management. Rep. Kim likely inquired about the management plan related to the significant prescription volume of Wegovy, which has garnered immense popularity since its launch in Korea. According to data from the Drug Utilization Review (DUR) system, approximately 400,000 prescriptions for Wegovy have been issued in the roughly eight months since its launch in October 2024, which translates to about 80,000 prescriptions per month. The non-reimbursed prescription price for Wegovy ranges from KRW 200,000 to 300,000 for the 0.25mg, 0.5mg, and 1.00mg doses, and over KRW 400,000 for the 1.70mg and 2.40mg doses. Related to this, Rep. Kim has raised the need for managing the side effects that arise from people who are not overweight or obese using these drugs for cosmetic purposes. The medical community has also pointed out that some doctors are prescribing Wegovy and other obesity drugs for cosmetic, rather than therapeutic, purposes. This practice, they argue, can lead to repeated prescriptions and illegal trading of excessively prescribed medicines on online platforms. To address these issues, the medical community is proposing that obesity treatments be brought under the National Health Insurance system, subjecting them to a public monitoring and management system. In response to these concerns, Rep. Kim asked HIRA for its plan to manage the side effects of Wegovy through reimbursement. However, HIRA's response was general and procedural. HIRA explained, "For a new drug to be listed for reimbursement, the pharmaceutical company must first apply for reimbursement to the Minister of Health and Welfare and the head of HIRA, along with the necessary documentation," and added, "Then, this is followed by HIRA will evaluate the drug's clinical utility and cost-effectiveness, followed by drug price negotiations with the National Health Insurance Service. Finally, a notification is issued by the Ministry of Health and Welfare." HIRA said, "Wegovy's manufacturer had not yet submitted a reimbursement application after its approval by the Ministry of Food and Drug Safety," and added, "If an application for this drug is submitted in the future, we will ensure that it is evaluated fairly and swiftly." Meanwhile, the decision on National Health Insurance reimbursement for new obesity drugs, such as Wegovy, will be based on South Korea's health insurance finances, reimbursement equity, and the cost-effectiveness of these drugs.
- Opinion
- [Reporter’s View] Be aware, be prepared
- by Eo, Yun-Ho Sep 03, 2025 06:10am
- Even if it seems premature, there is nothing wrong with being prepared. Following the Trump administration’s executive order on the Most-Favored-Nation (MFN) drug pricing policy, the deadline for major multinational pharmaceutical CEOs to submit their proposed drug price reduction plans is fast approaching—September 29. The MFN drug pricing policy seeks to adjust U.S. drug prices to match the lowest price among advanced countries. Initially, this standard was set to apply to Medicaid—health insurance for low-income patients—and gradually expand to Medicare, the public health insurance program. In short, U.S. drug prices will be aligned with the lowest prices found among developed nations. Experts warn that South Korea could become that benchmark country. Given the already heightened concern about “Korea passing,” this may further push multinational companies to avoid listing their drugs in Korea’s reimbursement system altogether. The U.S. pharmaceutical market is the largest in the world and accounts for nearly half of the global market share, which is more than 20 times than that of Korea’s. For multinational firms pursuing profit, Korea becomes a market they can easily abandon. The signs are already visible. Since Trump’s drug pricing policy was announced, some multinational pharmaceutical companies have withdrawn evaluation applications for new drug listings, while others have temporarily halted headquarters’ approval for such applications. Even products already listed have been affected: in one case, a company withdrew authorization, leading to deletion from the reimbursement list. It is time to revisit solutions that have long remained at the “proposal” stage, such as list price preserving mechanisms and structural improvements in expenditure. For drugs already listed, policymakers need to explore alternatives to continuous post-listing price cuts imposed through current pricing mechanisms. Admittedly, dual pricing is an inherently self-serving policy that undermines transparency. By concealing actual country-to-country pricing, it widens the ambiguity of drug costs. Yet, it is also an unavoidable choice for a government seeking to protect its own citizens. Within this dilemma, Korea must now make its own rational choice. There is a chance the scope of Trump’s “bomb” will shrink compared to its initial form. But it remains a possibility nonetheless—even if positive signals were received at the Korea–U.S. summit. Being aware and being prepared is essential.
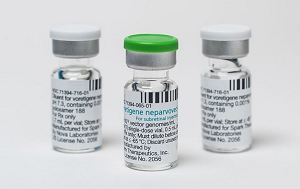
- Policy
- Luxturna shows signif improvement in 3 out of 6 patients
- by Lee, Tak-Sun Sep 03, 2025 06:09am
- Luxturna (voretigene neparvovec, Novartis), a one-shot gene therapy that costs approximately KRW 300 million, showed clinically significant changes set by reimbursement criteria in only half of the patients. As indicated in the performance evaluation results disclosed last year, the effectiveness was only 50%. The Health Insurance Review & Assessment Service (HIRA) disclosed the latest performance evaluation results for Luxturna on August 29. Luxturna is a gene therapy for patients with inherited retinal disease, administered as a single subretinal injection in each eye. The ceiling price for one vial is KRW 325.8 million, with a patient co-payment of approximately KRW 10.5 million per person. Due to its high cost, health authorities have a risk-sharing agreement (RSA) with three types of contracts (refund, expenditure cap, and performance-based refund) to manage the drug expenditure. The performance-based refund contract, in particular, requires a post-administration performance evaluation to adjust the refund rate. The detailed reimbursement criteria for this drug are as follows. 1. A clinical evaluation (light sensitivity, vision, visual field, etc.) must be performed before administration (within 90 days before the first eye's injection) and at 1-3 months, 12 months, and annually for up to 4 years after administration (after the second eye's injection if both eyes are treated). Objective records, such as medical charts, must be submitted. 2. Light sensitivity must be evaluated using a full-field light threshold test with white light. 3. A clinically significant change is defined as an improvement of 1 log unit (average value for both eyes) or more from the baseline in the full-field light threshold test. The first performance evaluation result was disclosed on October 31 of last year, following the reimbursement listing in February of that year. The first evaluation followed up on four patients 1-3 months after administration. The result showed that two patients had a significant improvement, while two did not, indicating a 50% success rate. The latest evaluation results are based on a total of six patient cases. The evaluation was conducted 1-3 months after administration for two patients and 12 months after for four patients. The results showed that one patient at 1-3 months and two patients at 12 months had a significant improvement. In contrast, it was evaluated that one patient at 1-3 months and two patients at 12 months did not meet the criteria for significant improvement. It means that half of the patients were successful, while the other half failed. Since Luxturna's performance evaluation will continue for up to 4 years, the efficacy is expected to be more accurately verified as more data accumulates. By adjusting the refund rate accordingly, the high cost of the drug can be controlled. HIRA is currently conducting performance evaluations for other high-cost drugs, including Kymriah, Zolgensma, and Qarziba.

- Policy
- Bill to mandate generic substitutions gains momentum
- by Lee, Jeong-Hwan Sep 03, 2025 06:09am
- On September 2, the ruling party introduced a bill to mandate generic (ingredient-based) prescriptions for medicines with unstable supply. The bill establishes the legal basis for “shortage drugs” and allows prescribing by ingredient name instead of brand name. The bill holds significance in that it amends the Pharmaceutical Affairs Act to define drugs frequently in shortage and sets out legal procedures to designate such through public-private consultations, and amends the Medical Service Act to legislate the legitimacy and standards for mandatory ingredient prescriptions. In other words, the bill enforces a law that stipulates that “drugs in such unstable supply that generic prescribing must be enforced” through deliberations by a public-private consultative body. Representative Jong-tae Jang of the Democratic Party of Korea, who spearheaded the amendments, has stated he will push for their swift passage. The participation of fellow Democratic Party representative Yoon Kim (a physician) and Rebuilding Korea Party Rep Sun-min Kim (also a physician) is expected to reduce obstacles in upcoming committee reviews. Specifically, the bill requires the Ministry of Health and Welfare (MOHW) to establish a Supply Management Committee for Shortage Drugs. After deliberation and resolution by this committee, the Minister of Health and Welfare will designate shortage drugs. The committee will have up to 30 members, including the Vice Minister of Health and Welfare as Chair, Deputy Commissioner of the Ministry of Food and Drug Safety (MFDS) as Vice Chair. The remaining 28 will consist of : ▲Senior officials from relevant central government agencies (appointed by Presidential Decree), ▲ representatives recommended by the Chair of the Korean Pharmaceutical Association, ▲ representatives recommended by the Korean Medical Association under Article 28 of the Medical Service Act, ▲ representatives recommended by the Korea Pharmaceutical and Bio-Pharma Manufacturers Association and other relevant industry groups, as well as ▲ experts with sufficient knowledge and experience. Thus, government officials, pharmacists, physicians, manufacturers, and academia will collectively decide which drugs require mandatory generic prescribing due to unstable supply. The Minister of Health and Welfare will also have dedicated staff and budget authority to designate and de-designate shortage drugs, monitor supply conditions, and implement distribution improvement measures. The bill also legalizes distribution interventions alongside generic prescribing. If supply is deemed significantly disrupted, or upon request from another central administrative agency head, the Minister may—after committee review—order measures to improve distribution regarding sales outlets, procedures, volumes, and conditions. Pharmacies, medical institutions, wholesalers, and other designated entities will be legally obligated to comply with these measures. However, the Minister must first consult with the Minister of Strategy and Finance and the Chair of the Fair Trade Commission before issuing such orders. The MOHW Minister will also build and operate a shortage drug management system. This will allow requesting and collecting information necessary for distribution control, which includes production, shipments, sales, prescriptions, and dispensing data, from manufacturers, importers, wholesalers, pharmacies, and medical institutions. This effectively grants the MOHW authority over the entire supply chain, but also increases the Ministry’s accountability when shortages occur. The Minister may also designate certain shortage drugs as “emergency production/import drugs”, subject to committee review, which empowers the Minister to order manufacturers to produce or import the drugs. The Medical Service Act amendment stipulates that physicians and dentists must prescribe designated shortage drugs by generic name, not brand name. Violation will result in up to 5 years imprisonment or a fine up to KRW 50 million. This significant level of criminal penalty is expected to draw strong opposition from the Korean Medical Association and the broader medical community.
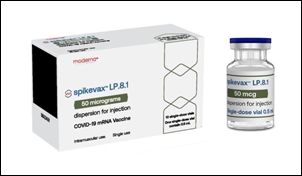
- Company
- Moderna’s latest variant-targeted COVID-19 vaccine approved
- by Whang, byung-woo Sep 03, 2025 06:08am
- Moderna Korea announced on the 1st that its LP.8.1 variant-targeted COVID-19 vaccine, ‘Spikevax LP Inj’, has been approved by the Ministry of Food and Drug Safety (MFDS). Spikevax LP has been confirmed to induce broad cross-immune responses against currently circulating variants, including the LP.8.1 strain, and it is authorized for use in adolescents aged 12 and above as well as adults. Moderna plans to supply the newly approved vaccine in time for the government’s 2025–2026 seasonal immunization program, which begins in October. The company noted that its COVID-19 vaccines have demonstrated strong immune effect and safety in large-scale Phase III clinical trials and extensive real-world evidence (RWE). A key feature is that elderly individuals aged 65 and above showed immune responses comparable to those in younger adults. Also, regardless of which vaccine type had been administered previously, the Moderna vaccine showed high immunogenicity when used as a subsequent dose. A domestic study conducted by the Korea Disease Control and Prevention Agency (KDCA) also confirmed that Moderna’s vaccine recorded the lowest breakthrough infection rate among the vaccines used in the early stages of the pandemic. According to the National Immunization Program guidelines, Spikevax LP will be provided free of charge to high-risk groups, including seniors aged 65 and over and residents of long-term care facilities. The Korean Society of Infectious Diseases has stressed that immunity acquired through infection or vaccination wanes over time, and with the emergence of new variants, periodic updated COVID-19 vaccination for high-risk groups is recommended. The LP.8.1 series vaccines supplied this season have already been recommended for use by the World Health Organization (WHO), the European Medicines Agency (EMA), and the U.S. Food and Drug Administration (FDA). Based on recommendations from its Vaccination Expert Committee, the KDCA decided to adopt the LP.8.1 vaccine, which demonstrated stronger neutralizing antibody responses compared to last season’s JN.1-based vaccines. Moderna is the only company manufacturing mRNA COVID-19 vaccines in Korea, through its partnership with Samsung Biologics, and continues to ensure a stable vaccine supply via ongoing cooperation with Boryung Biopharma. Sang Pyo Kim, General Manager of Moderna Korea, said, “COVID-19 remains a threat to high-risk groups, with hospitalizations on the rise for 7 consecutive weeks. Moderna is committed to delivering updated COVID-19 vaccines that address the latest variants in a timely manner, ensuring that people can be vaccinated safely.”
- Company
- 'Prevenar 20' now available at general hospitals
- by Eo, Yun-Ho Sep 02, 2025 06:11am
- Product photo of Prevenar 20 PFS The pneumococcal conjugate vaccine 'Prevenar 20,' which will soon be included in the National Immunization Program (NIP), is becoming available for prescription at general hospitals. According to industry sources, Pfizer Korea's Prevenar 20 has passed the drug committees (DC) of tertiary general hospitals, including Samsung Medical Center, Seoul National University Hospital, Asan Medical Center in Seoul, and Sinchon Severance Hospital, and medical institutes, including Kangnam Sacred Heart Hospital, Kyung Hee University Hospital at Gangdong, Pusan National University Hospital, Seoul National University Bundang Hospital, Ajou University Hospital, Pusan National University Yangsan Hospital, and Chungnam National University Hospital. Several of these hospitals, including Seoul National University Hospital, have approved codes for adult patients only. However, as Prevenar 20 will be included in the NIP effective October, more hospitals are expected to have pediatric vaccines. Prevenar 20 is a pneumococcal conjugate vaccine that was approved by the Ministry of Food and Drug Safety (MFDS) on October 31, 2024. Compared to the 13-valent vaccine, Prevenar 20 has added seven additional pneumococcal serotypes. Among domestically approved pneumococcal conjugate vaccines, it contains the most serotypes. In addition to existing serotype components in the 13-valent vaccine, Prevenar 20 contains seven additional serotypes (serotypes 1, 3, 4, 5, 6A, 6B, 7F, 8, 9V, 10A, 11A, 12F, 14, 15B, 18C, 19A, 19F, 22F, 23F, 33F). It can be used to prevent invasive diseases and pneumonia in all ages, including infants aged six weeks and above. Pneumococcus is a major bacterial pathogen that causes various diseases in infants and young children, including otitis media, pneumonia, and meningitis. Vaccination is crucial, especially as it can cause life-threatening invasive pneumococcal disease (IPD) in immunocompromised children. Despite South Korea's NIP supporting the 23-valent pneumococcal polysaccharide vaccine (PPSV23) for adults aged 65 and older, this age group has the highest incidence of invasive pneumococcal disease (IPD). From September 2014 to mid-November 2023, a total of 3,734 cases of IPD were reported. The incidence rate for adults aged 65 and over was 32.1 cases per 100,000 people, accounting for 54.8% of all cases. This rate is the highest among all age groups under 65 years old. The inclusion of Prevenar 20 in the NIP was decided after a comprehensive review by the Korea Expert Committee on Immunization Practices (KECIP) on the vaccine's safety, immunogenicity, and cost-effectiveness. Meanwhile, Korea Vaccine is responsible for the domestic promotion of pediatric Prevenar 20, while Chong Kun Dang handles the promotion of the adult vaccine.