- LOGIN
- MemberShip
- 2026-06-14 08:30:06

- Company
- Patient & Consumer Alliance says "Structural reform needed for drug pricing reform"
- by Son, Hyung Min Mar 31, 2026 08:45am
- Patient & Consumer Alliance for Healthcare Rights (PCA) provided both positive evaluations and concerns over structural limitations regarding the recent drug pricing system reform. The drug price cut is a significant change; However, they argue that a breakthrough reform remains difficult as long as the market remains centered on rebate-driven structures.On the 27th, the PCA stated as such after the Health Insurance Policy Review Committee approved the "Measures to Improve the National Health Insurance Drug Pricing System" the previous day.The Patient & Consumer Alliance for Healthcare Rights (PCA) was officially launched on the 24th joined by four public citizens and patient organizations, including the Consumers Union of Korea, the Korea Alliance of Patient Organizations, Citizens' Movement for Consumers, and the Korean Organization for Rare Diseases, with the goal of shifting the current "government-led and provider-centric" medical structure toward a patient- and consumer-centered one (photo= Korean Organization for Rare Diseases).Previously, the Ministry of Health and Welfare (MOHW) finalized a plan to lower the drug price calculation rate for generics and off-patent drugs from the current 53.55% to 45%. The PCA evaluated this measure as "meaningful progress, marking the first reform of the generic drug pricing structure in 14 years."However, the PCA stated that price reductions alone are insufficient to eradicate the practice of rebates.The PCA stated, "Drug prices are not commensurate with rebates and that rebate practices will persist as long as the unfair competitive structure across licensing and distribution remains intact."They also raised concerns about the tiered price adjustment structure and the newly introduced "New Innovative Pharmaceutical Company" system included in the reform.According to the PCA, "The grace period of up to 10 years delays market restructuring," adding, "New Innovative Pharmaceutical Company" system lacks clear criteria and evaluation methods, potentially allowing companies without innovative capabilities to be incorporated into the support system.Furthermore, the PCA identified potential issues with the government's policies supporting the pharmaceutical and biotech industries. They argued that if this continues, with savings from price cuts being reinvested in industrial support, it could result in using taxpayer money to sustain companies with uncertain competitiveness.The PCA further stated, "The scale and performance of investments into pharmaceutical R&D over the 26 years since the separation of prescribing and dispensing have never been fully disclosed to the public," adding, "Discussions on additional support should be provided only after independent performance evaluations are made public."Issues were raised regarding the sales structures of certain companies within the Korean pharmaceutical market.The PCA pointed out, "There may be so-called bogus pharmaceutical companies that maintain market presence through rebate-driven sales via Contract Sales Organizations (CSOs) without possessing production facilities or research capabilities," and argued that "These companies undermine the competitive foundation for legitimate companies."The PCA stated, "If this structure persists even after lowering drug prices, companies relying on rebates, rather than those with true competitiveness, will maintain their market status," and adding, "The key is the normalization of the competitive order rather than just price reduction."The PCA stressed that government support is necessary to improve the pharmaceutical market structure. They called for the immediate launch of a 'Pharmaceutical Market Fair Trade Task Force (TF)' involving the Ministry of Health and Welfare, the Ministry of Food and Drug Safety, and the Fair Trade Commission to promote: ▲Eradicating rebates and strengthening CSO management ▲Establishing exit criteria for pharmaceutical companies without production capabilities ▲Disclosing performance results of financial support for pharmaceutical R&D.The PCA stated, "Drug price reduction is only the beginning, and as long as the rebate structure remains, patients may not receive benefits," adding, "The government must prioritize creating a foundation for fair competition before returning financial savings to the industry."The PCA concluded, "We hope this pricing reform serves as a starting point for reform leading to a fair pharmaceutical market," and stated, "The alliance would continuously monitor the implementation process." The PCA emphasized that government support is necessary to improve the pharmaceutical market structure.

- Opinion
- "Joint bioequivalence licensing system is driving abundant generics"
- by Lee, Jeong-Hwan Mar 31, 2026 08:45am
- Director Lee Jae HyunWhile the government announced plans to divert from the multi-item generic structure and improve the pharmaceutical industry’s trend by focusing on new drugs through drug pricing reforms, criticism has emerged that there is a need to innovate by diagnosing blind spots in the "approval system" beyond pricing alone.In Korea, it has been pointed out that the so-called '1+3 system,' under which the government grants marketing authorization for generics based on a single joint bioequivalence (BE) study conducted by one contract manufacturer and up to three consigned pharmaceutical companies, must be discarded.Furthermore, it has been suggested that, for the long-term development of the domestic pharmaceutical industry, the government must make a policy decision to allow South Korea's world-class clinical physician workforce to enter new drug development.In an interview on the 29th at the Sungkyunkwan University Center for Pharmaceutical Regulatory Sciences in Yeongdeungpo, Seoul, Director Lee Jae Hyun (Professor at Sungkyunkwan University College of Pharmacy) emphasized, "Why we are seeing hundreds of generics for a single active ingredient today is not due to high drug prices, but because of the joint bioequivalence licensing system."Regarding the '1+3 system,' which grants generic marketing authorization even to companies that did not conduct in-house BE tests but instead outsourced them to a contract manufacturer, Lee evaluated it as a "policy with no global precedent."Lee questioned whether a company can truly be called a "pharmaceutical company" if it simply purchases BE test data conducted by others, obtains authorization for identical twin generics, and profits by releasing them to the market after merely changing the brand name and packaging.Lee noted, "Although it was reduced to '1+3' (from the previous unlimited structure), the concept of joint BE licensing is preposterous. Even a '1+1' system, where only one consigned license is granted per contract manufacturer, makes no sense. Lee stated, "This is the fundamental cause of generic proliferation, which undermines the fundamentals of regulatory science. Why do drugs with identical shapes, ingredients, and dosages have different manufacturers, brand names, and packaging?"Lee emphasized, "If one set of BE data exists, marketing authorization should be granted only to that specific pharmaceutical company. As long as this joint BE licensing system remains in place, a structure in which generic substitution is hindered, and generics proliferate, will persist. We should not allow companies to outsource production just by hanging up a 'pharmaceutical company' sign. We must abolish these mismanaged licensing and joint BE policies."Lee explained, "If the licensing policy is corrected, drug price management can also be designed much more rationally. A policy could be designed that grants 'branded generic' rights only to the original and the first generic, while all other generics are authorized as 'no-brand generics' using the ingredient name rather than a brand name," adding, "Policies to increase generic utilization could become possible, and the controversy over INN (International Nonproprietary Name) prescribing would disappear, as the product name itself would be authorized as the ingredient name."Director Lee also believes that to develop Korea-Blockbuster new drugs, Korea’s abundant clinical physician workforce must be funneled into new drug development, where basic science is essential.While the stages of new drug development include discovery, demonstrating efficacy and safety through clinical trials, and obtaining marketing authorization, Lee suggests establishing policies to recruit physicians, one of Korea's greatest strengths, in the actual development of new drugs.Director Lee suggested, "New drugs are not developed by government pricing policies or pharmaceutical companies. They emerge from the advancement of a country's life sciences and the continuous increase in research on new drug substances," adding, "I believe the cultivation of talent for developing new drugs ultimately lies in the utilization of the physician workforce. Implementing administration that actively utilizes Korea's globally powerful resource in the clinical trial field is the shortcut to becoming a powerhouse in new drugs."Lee stated, "We must allow doctors to engage in new drug activities through policies such as designating university hospitals as new drug development centers, providing military service benefits to participating doctors, and expanding national-level support for new drug ventures and startups within those institutions. It is difficult to develop new drugs solely within the pharmaceutical industry or regulatory science. A policy decision is needed to use doctors as the foundation for blockbuster new drugs."Lee added, "Furthermore, we need to consider amending the outdated Pharmaceutical Affairs Act. Korea's Pharmaceutical Affairs Act has not been changed from a generic-centered legal structure since the 1950s. While a pharmaceutical company is defined as one that manufactures and produces drugs, there is no system where a new drug developer can receive product approval as a pharmaceutical company."Lee concluded, "We need to create a Pharmaceutical Affairs Act to manage pharmaceutical personnel, have a Drug Safety Management Act handle synthetic drugs and new drug approvals, and separate biological products from synthetic drugs through a "Biopharmaceutical Act" or an "Advanced Biologics Act," adding, "This is a matter that requires a shift in the government's regulatory paradigm. It is time to consider shifting the manufacturer-centered Pharmaceutical Affairs Act into a basic act and separately operating laws for drug distribution management, synthetic drug licensing, and biological products."

- Policy
- Premiums for incrementally modified drugs remain uncertain
- by Jung, Heung-Jun Mar 31, 2026 08:45am
- As the government has finalized the overall framework of the drug pricing system reform, discussions are expected to move into detailed areas, including premiums for incrementally modified drugs.The core of this drug pricing system reform is the innovative level of companies. However, decisions regarding product-level premium measures, such as those for incrementally modified drugs, have been left out.According to industry sources on the 27th, since the premium rate for incrementally modified drugs was not clearly determined at this Health Insurance Policy Deliberation Committee meeting, follow-up discussions are expected to continue.The reform plan discussed at the Health Insurance Policy Deliberation Committee meeting last November had tentatively decided to maintain the current premium system for “incrementally modified drugs, incrementally modified combination drugs, and biosimilars.”However, this wording was deleted at the latest HIPDC meeting. With the addition of a new “quasi-innovative company” category, the drug price premium scheme has been significantly revised. The disclosed premium preferential measures consist solely of a 60% premium for innovative drugs, a 50% premium for quasi-innovative drugs, and preferential treatment for pharmaceutical companies and drugs that ensure supply stability.Under the current pricing calculation system, incrementally modified drugs receive premiums on top of the base price. For salt/formulation changes after original patent expiry, the price is set at 70% with a premium from the base price (53.55%). The price is set at 77% with a premium from the base price (58.9%). The premium is generally applied for 1 year after listing, but can be extended up to 3 years if there are three or fewer generic manufacturers listed for reimbursment.The key question is whether the government will maintain these premium rates. Since the base calculation rate has been lowered, there is a possibility that the premium rates will also be readjusted. If the government attempts to lower the premium rates for incrementally modified drugs in line with the reduced base rate, strong industry backlash is expected. The prevailing view in the industry is that if the government attempts to adjust the premium rate as well, it will dampen the motivation to develop incrementally modified drugs.There is also the possibility of reforming premiums for incrementally modified combination drugs. Currently, their price is calculated as the sum of 53.55% of each component drug’s pre-patent-expiry price. In this case, innovative pharmaceutical companies receive preferential treatment with a 68% sum, while general pharmaceutical companies receive a 59.5% sum.Given that drug price premium tiers are currently divided into innovative, quasi-innovative, and non-innovative categories, changes are needed in the calculation rates for incrementally modified drugs as well.In particular, since the “quasi-innovative” category is a newly established preferential pricing bracket, discussions must also address whether to apply differential premium rates within this category.Industry insiders agreed that, as the basic calculation rates and drug price premiums had not been finalized until now, concrete discussions must begin immediately.A pricing manager at a pharmaceutical company stated, “Until now, the focus was on establishing the overall framework, so detailed discussions were not possible. Now, discussions on incrementally modified drug premium rates must begin.”Another industry official added, “The content announced at this Health Insurance Review and Assessment Service (HIRA) meeting only covers the broad framework, so significant detailed adjustments are needed. The same applies to the premium rate for incrementally modified drugs. While each pharmaceutical company may focus on different aspects of the reform, since the base calculation rate has decreased, it is necessary to ensure that premiums are maintained as much as possible.”

- Opinion
- ‘Revisiting Xeljanz safety concerns based on accumulated long-term data’
- by Son, Hyung Min Mar 31, 2026 08:45am
- Eun-mi Song, Professor of Gastroenterology and Hepatology, Ewha Womans University Seoul HospitalAs treatment strategies for ulcerative colitis shift toward maintaining long-term remission, the criteria for selecting treatment options are also changing.In particular, as Janus kinase (JAK) inhibitors establish themselves as key treatment options alongside biologics, safety concerns surrounding Pfizer’s ‘Xeljanz (tofacitinib),’ including major adverse cardiovascular events (MACE) and thrombosis, have been consistently raised.Amid this context, as results from domestic cohort studies involving Korean patients accumulate, there is a growing movement to reassess its safety in real-world clinical settings.Professor Eun-mi Song of the Department of Gastroenterology at Ewha Womans University Seoul Hospital, who led this study, recently told Daily Pharm, “Initially, there were significant concerns about side effects due to the mechanistic characteristics of JAK inhibitors. Looking at actual clinical data, contrary to expectations, the safety profile is comparable to that of existing biologics.”Ulcerative colitis is a disease characterized by chronic inflammation of the colon's mucosa, with recurring symptoms including diarrhea, bloody stools, and abdominal pain. Unlike acute colitis, it is a chronic condition with no clear cause that involves repeated cycles of remission and relapse, requiring many patients to continue treatment for the rest of their lives.The number of patients in Korea is also rapidly increasing. Due to Westernized dietary habits and environmental changes, prevalence is rising, particularly among younger patients aged 20–40. As the number of patients in this age group increases, which is highly active in society, the need for long-term disease management also grows.At the same time, the treatment landscape is evolving. While a step-up strategy, gradually increasing treatment intensity, was previously the norm, an ‘accelerated step-up’ strategy, which adjusts treatment intensity more quickly based on the patient’s condition, is now being applied in clinical practice. Treatment goals are also evolving beyond simple symptom relief toward the fundamental suppression of inflammation, such as achieving endoscopic remission.The problem lies in the high recurrence rate. Since more than 80% of patients experience recurrence and some progress to severe disease, the continuity of treatment, which maintains stable suppression of inflammation even after initial remission, is identified as a key factor determining long-term prognosis.Accordingly, efforts are ongoing to establish strategies for maintaining long-term remission while also validating drug safety in real-world clinical settings.In particular, JAK inhibitors have faced persistent safety concerns since their introduction, including risks of MACE, thrombosis, infections, and malignancies.However, it has been pointed out that these risks were primarily derived from data on rheumatoid arthritis, which involves a large proportion of elderly patients, and that there are limitations to applying them directly to the ulcerative colitis patient population, which has a relatively high proportion of younger patients.Furthermore, in actual clinical practice, the patterns of adverse reactions may vary depending on patient comorbidities, age, and concomitant therapies, reinforcing the need for the collection of real-world data from domestic patient populations.Against this backdrop, a large-scale population-based cohort analysis was conducted in Korea. Using data from the National Health Insurance Service (NHIS), the study compared the risk of serious adverse events (SAEs) between the Xeljanz group (521 patients) and the TNF inhibitor group (1,295 patients) in patients with moderate-to-severe ulcerative colitis from May 2019 to April 2022.Analysis revealed that the overall incidence rate of SAEs was 4.41 per 100 person-years in the Xeljanz group and 5.33 in the TNF inhibitor group, showing no statistically significant difference between the two treatment groups. In particular, no differences were observed between the groups in the risk of thromboembolism, opportunistic infections such as herpes zoster and tuberculosis, or malignancies.Professor Song stated, “The occurrence of complications is influenced more by individual risk factors, such as the patient’s age or underlying conditions, than by the drug itself. If treatment and monitoring are conducted in conjunction with consideration of each patient’s risk level, Xeljanz is a viable long-term treatment option.”Q. Given the reimbursement criteria, the top-down approach seems ideal, but the step-up approach still predominates in real life.In Korea, ulcerative colitis treatment is moving toward an ‘accelerated step-up’ approach, which is a practical compromise between top-down and traditional step-up strategies. This involves closely monitoring patient response and rapidly escalating to more potent therapies when initial treatments are insufficient, enabling early remission.In the past, treatment typically began with 5-aminosalicylic acid (5-ASA) agents, followed by sequential use of immunomodulators in a stepwise approach; however, in recent practice, steroids or immunomodulators are used from the outset in patients with severe symptoms. In particular, if the disease continues to worsen despite this initial intervention, biologics or small-molecule agents (JAK inhibitors) such as Xeljanz are introduced early on, in accordance with domestic health insurance reimbursement criteria.Q. When considering switching, what specific criteria are used to make the change?Disease severity is the primary factor. Physicians assess symptom severity, endoscopic inflammation, and laboratory results to evaluate disease status. The first criterion is selecting the treatment with the highest expected efficacy based on severity, followed by safety considerations.The criteria for selecting ulcerative colitis treatments have evolved to comprehensively consider not only the patient’s clinical characteristics but also safety in relation to comorbidities, as well as the patient’s individual preferences and lifestyle patterns. While treatment options were limited in the past, the recent introduction of new drugs with diverse administration routes and schedules has made it possible to design sophisticated treatment plans tailored to each patient’s specific situation.Q. How was the Xeljanz cohort study conducted?This large-scale, population-based cohort study of Korean ulcerative colitis patients was designed to directly compare Xeljanz with TNF inhibitors, which have been in clinical use for a relatively long period and are considered to have an established safety profile, as the control group.The study results confirmed that the safety profile of Xeljanz is comparable to that of existing biologics, namely TNF inhibitors. Initially, due to its mechanism of action, it was anticipated that the Xeljanz group would have a relatively higher risk of viral infections or thrombosis, however, actual analysis revealed no statistically significant difference in the incidence of serious adverse events between the two treatment groups.However, a major limitation of this study is that specific data on initial drug dosing were not obtained during the study, preventing precise analysis of dose-dependent safety and efficacy differences. Previous studies, such as ORAL Surveillance, have already suggested that dosage differences in JAK inhibitors can have a significant impact on safety outcomes, and there remains a possibility that this study could also reveal differences in clinical outcomes based on dosage.Q. Despite the periodic release of safety data on Xeljanz, concerns regarding risks still persist.Even among healthcare professionals, there is a vague fear of complications when prescribing JAK inhibitors like Xeljanz. However, clinical data reported domestically and internationally to date show that these concerns do not translate into actual risks. In conclusion, it has been confirmed that effectively controlling the inflammatory state early on with Xeljanz, which has a potent and rapid effect, actually contributes to reducing the risk of disease-related complications and ensuring long-term patient safety.Furthermore, compared to Western populations, the absolute probability and incidence of thrombosis in Asian patient groups have been observed to be relatively lower. While Western populations exhibit higher thrombosis rates due to factors such as larger body frames and a higher proportion of obese individuals, Asian populations show a similar trend of increased risk compared to the general population, yet the absolute number of cases tends to be lower.Previous safety warnings regarding JAK inhibitors were primarily based on data from rheumatoid arthritis, which involves a large number of elderly patients in their 50s and 60s. However, since the ulcerative colitis patient population consists mostly of younger individuals, the risks of thrombosis and other complications, which were raised in the rheumatoid arthritis data dominated by elderly patients, were found to be relatively lower.Q. Do clinical experiences in actual prescribing practice show similar patterns to study findings in terms of efficacy and safety?Combined results from domestic multicenter studies and real-world clinical experience indicate that the safety concerns raised during the early stages of the introduction of JAK inhibitors, including Xeljanz, do not pose a significant problem in real-world clinical settings. In particular, regarding herpes zoster, which was expected to carry a high risk based on the mechanism of action, no serious safety issues as previously feared emerged, thanks to thorough preemptive vaccination of healthcare providers and close monitoring. On the contrary, the greatest strength of Xeljanz perceived in clinical practice was its very rapid and potent efficacy, providing immediate therapeutic benefits to patients in urgent need of rapid symptom improvement.Q. What are the limitations of the current treatment environment, including reimbursement?While Western practice emphasizes top-down strategies for improved long-term outcomes, Korea faces practical constraints regarding the application of early, potent treatment due to the National Health Insurance system and financial limitations.Thus, greater flexibility in enabling early use of potent therapies, especially for severe patients, is considered essential for improving treatment outcomes. Furthermore, recent accumulated data have demonstrated that switching between different JAK inhibitors yields clinically significant efficacy. Consequently, it is anticipated that switching between different JAK inhibitors, even if a patient shows an inadequate response to a specific JAK inhibitor, may provide meaningful clinical benefits, offering additional options for patients who do not respond adequately to a specific agent.Q. What is the clinical significance of JAK inhibitors in ulcerative colitis?Despite their relatively recent introduction, small-molecule therapies have become a core pillar, playing a central role in the treatment of ulcerative colitis. While there was once a vague apprehension regarding these treatments even among clinicians, the experience and data shared by professors who have prescribed drugs such as Xeljanz in actual clinical practice confirmed that the risk of complications, which had been a concern, was lower than expected and manageable.In particular, for moderate-to-severe patients who struggled with the burden of even daily outings due to recurring cycles of symptom improvement and flare-ups, oral small-molecule agents, which offer high convenience, have become a practical alternative that dramatically improves quality of life. With the recent expansion of available treatment options to 3 or more, led by the introduction of Xeljanz, and ongoing new drug development, it is crucial for patients to maintain hope and work closely with healthcare professionals to establish a treatment strategy optimized for their individual needs in order to maintain long-term remission.
- Company
- Opdivo reattempts reimb expansion for first-line liver·lung cancer
- by Eo, Yun-Ho Mar 31, 2026 08:45am
- Immunotherapy Opdivo is set to reattempt reimbursement listing for liver and lung cancer in Korea.According to industry sources, Ono Pharmaceutical Korea’s PD-1 inhibitor Opdivo (nivolumab) is expected to be submitted to the Health Insurance Review and Assessment Service (HIRA) Cancer Disease Deliberation Committee in April.Last October, Opdivo failed to secure coverage criteria from the Cancer Disease Deliberation Committee for first-line treatment of hepatocellular carcinoma and non-small cell lung cancer. At that time, reimbursement criteria were only established for pleural mesothelioma.Ono immediately resubmitted applications for reimbursement expansion for the two indications, which will be reviewed again next month.Opdivo is essentially one of the earliest immuno-oncology drugs introduced, alongside Keytruda (pembrolizumab). However, it has remained a non-reimbursed indication for first-line NSCLC for an extended period. Discussions on reimbursement for first-line NSCLC have been ongoing since 2021.In hepatocellular carcinoma, Opdivo is indicated in combination with the CTLA-4 inhibitor Yervoy (ipilimumab). This combination regimen has demonstrated the longest survival data among first-line treatment options for hepatocellular carcinoma.The Opdivo + Yervoy combination showed a median overall survival (OS) of 23.7 months in the phase III CheckMate-9DW trial, which included patients with unresectable or advanced hepatocellular carcinoma who had not received prior systemic therapy. This represents a 21% reduction in the risk of death compared to the control group treated with ‘Lenvima (lenvatinib)’ or ‘Nexavar (sorafenib),’ which showed a median OS of 20.6 months.It remains to be seen whether Opdivo, which has faced a rocky road from its initial listing to the reimbursement expansion, will be able to expand its prescription scope this time.
- Company
- "Do we really need BE testing for already-listed generics?"
- by Chon, Seung-Hyun Mar 31, 2026 08:45am
- The pharmaceutical industry is assessing potential losses from price cuts for already-listed generic drugs. Substantial losses are expected as the government has officially announced that the new, lower price calculation rates will apply to already-listed generic drugs.With the standardized price calculation rate lowered and the highest price requirements expanded, generics without direct bioequivalence (BE) testing are expected to see their prices drop by more than 20%. There may be instances where companies rush to conduct bioequivalence studies on already approved products to avoid price reductions.On the 26th, the Ministry of Health and Welfare (MOHW) finalized the "Measures to Improve the National Health Insurance Drug Pricing System" during a meeting of the Health Insurance Policy Review Committee, confirming that existing drugs will be adjusted based on the revised calculation standards.Under the reformed system, the price for both off-patent original drugs and generics will decrease from 53.55% to 45% of the new drug's pre-patent-expiry price. The MOHW plans to categorize already-listed drugs into groups based on whether they were listed before or after 2012 and gradually adjust them to the 45% level. Both generics and the off-patent originals with listed generics are subject to these cuts.To maintain drive for new drug development, the MOHW will grant temporary exceptions for "Innovative" and "New Innovative" pharmaceutical companies. Under this scenario, Innovative companies will have their generic price calculation rate set at 49% for four years, while "New Innovative" companies will receive a rate of 47% for three years before eventually reaching the 45% criteria. Companies that do not fall into these categories will also face price cuts over a four-year period, likely dropping to 49% next year, 47% in 2028, and finally 45% in 2029.Under the reformed system, the price for both off-patent original drugs and generics will decrease from 53.55% to 45% of the new drug's pre-patent-expiry price; a generic failing one requirement will drop to 36%, and one failing both will drop to 28.8%.Pharmaceutical companies are primarily concerned about the loss resulting from these price adjustments on existing products. For instance, if a product with annual sales of KRW 10 billion has its price reduced from 53.55% to 45%, it mathematically results in an annual revenue decrease of KRW 1.6 billion. Effectively, KRW 1.6 billion in operating profit per product would evaporate. The price cut range is even greater if the top-tier price requirements, such as performing direct BE studies and using registered drug substances (DMF), are applied to these already-listed generics.Under the reformed system, the penalty for failing to meet top-tier price requirements will expand from 15% to 20%. Since July 2020, a system was introduced where generics could only receive the 53.55% maximum price if they met both the direct BE study and DMF requirements. For every requirement not met, the ceiling price dropped by 15%; failing both resulted in a 27.75% reduction. Currently, under the 15% penalty rule, a generic failing one requirement drops to 45.52%, and failing both drops to 38.69%.However, applying the new 45% requirement and the increased 20% penalty means that a generic failing one requirement will drop to 36%, and one failing both will drop to 28.8%. The price for a generic failing one requirement will be 20.9% lower than current levels, while those failing both will see a 25.6% decrease. Pharmaceutical companies with generics that have not undergone BE studies would have to endure a 20.9% price cut.An industry official stated, "For consigned generics, the price cut could be mitigated by performing a BE study, forcing companies to calculate whether the cost of the study outweighs the benefit of maintaining a higher price."Consequently, companies have begun reviewing the profitability of their generic portfolios. For products where profitability would be significantly damaged by the 20.9% cut, they may seek strategies to minimize losses, such as initiating late-stage BE studies.The industry is concerned that the confusion seen during the two rounds of price re-evaluations for approximately 8,000 generic items in September 2023 and March last year may recur.On September 5, 2023, the prices of 7,355 generic items were reduced by up to 28.6%, which was the first result of the generic price re-evaluation project launched in 2020. At that time, the MOHW announced that generics failing top-tier requirements could maintain their previous prices if they submitted proof of BE studies and DMF use by the end of February 2023. This policy was intended to apply the new 2020 pricing system to previously listed generics.At that time, most of the 7,355 items were hit with a 15% reduction, largely because they lacked BE studies. A total of 145 items saw cuts exceeding 20%, and 125 items saw cuts exceeding 27% because they failed both requirements, resulting in price drops approaching 30%. A total of 179 companies suffered losses from the first round of price cut. Korea Huons had 154 items affected, while Hana Pharm and Daewoong Bio saw cuts to 122 and 104 items, respectively.In March 2024, the second round of re-evaluations resulted in price cuts of up to 27.9% for 948 items. These additional cuts targeted sterile preparations like injections that were newly classified as subjects for equivalence testing.At that time, 125 items containing Artemisia ethanol soft extract saw prices drop by an average of 14.5% and a maximum of 27.4%. Artemisia extract is a natural product-based medicine used for gastric lesions. Stillen is the original product. Because it is difficult to prove equivalence for herbal medicines via traditional blood concentration levels, most of these generics could not fulfill the BE study requirement and were forced to accept the cuts.Drug prices were reduced for 94 generic items of Stillen and 31 generic Stillen 2X. These generic products of Stillen and Stillen 2X had been authorized based on comparative dissolution and comparative disintegration tests rather than bioequivalence (BE) studies. Because they failed to conduct BE studies (one of the requirements for the highest generic drug price) the prices of all these generic products were lowered. Among the 125 items subject to the price reduction, 108 saw their prices decrease by 15% due to failure to meet the BE study requirementPharmaceutical companies gave up conducting BE studies and were forced to accept price cuts, arguing that it is difficult to prove equivalence through BE studies, which compare blood concentrations of active ingredients, because of the specific nature of herbal preparations.There are concerns among pharmaceutical companies that efforts to conduct BE studies for price maintenance may resurface, as generic drug prices will drop even further following the reform of the drug pricing system.While companies previously gave up on BE studies for low-volume products and accepted the 15% cut, the higher 20.9% penalty and lower base price may trigger a vicious cycle of wasteful spending to protect revenue.In fact, during the previous re-evaluation, the rush to conduct BE studies for the sake of price maintenance led to significant social costs.According to the Ministry of Food and Drug Safety (MFDS), BE study approvals rose from 178 in 2018 to 323 in 2020, an 81.4% increase in two years, and reached 505 in 2021, nearly triple the number from three years prior. This phenomenon involved companies conducting new BE studies on products already on the market, then switching from "consigned manufacturing" to "in-house manufacturing" via permit changes to satisfy the "direct BE" requirement and evade price cuts.Once the re-evaluation ended, BE approvals returned to a downward trend, dropping to 296 in 2022, 229 in 2023, and 197 in 2024, returning to levels seen six years ago.Pharmaceutical companies have criticized these mandatory BE studies for already-listed drugs as a "waste of money."They argued that it is exhausting to spend upwards of KRW 500 million per BE study on drugs whose safety and efficacy have already been proven to meet a pricing requirement. Some companies have collectively spent billions of won on these efforts.An industry representative commented, "We are currently calculating the revenue impact and price reduction rates for products undergone price cuts during the last re-evaluation because BE studies were not conducted. We are devising strategies to minimize losses as the new pricing system is implemented."
- Policy
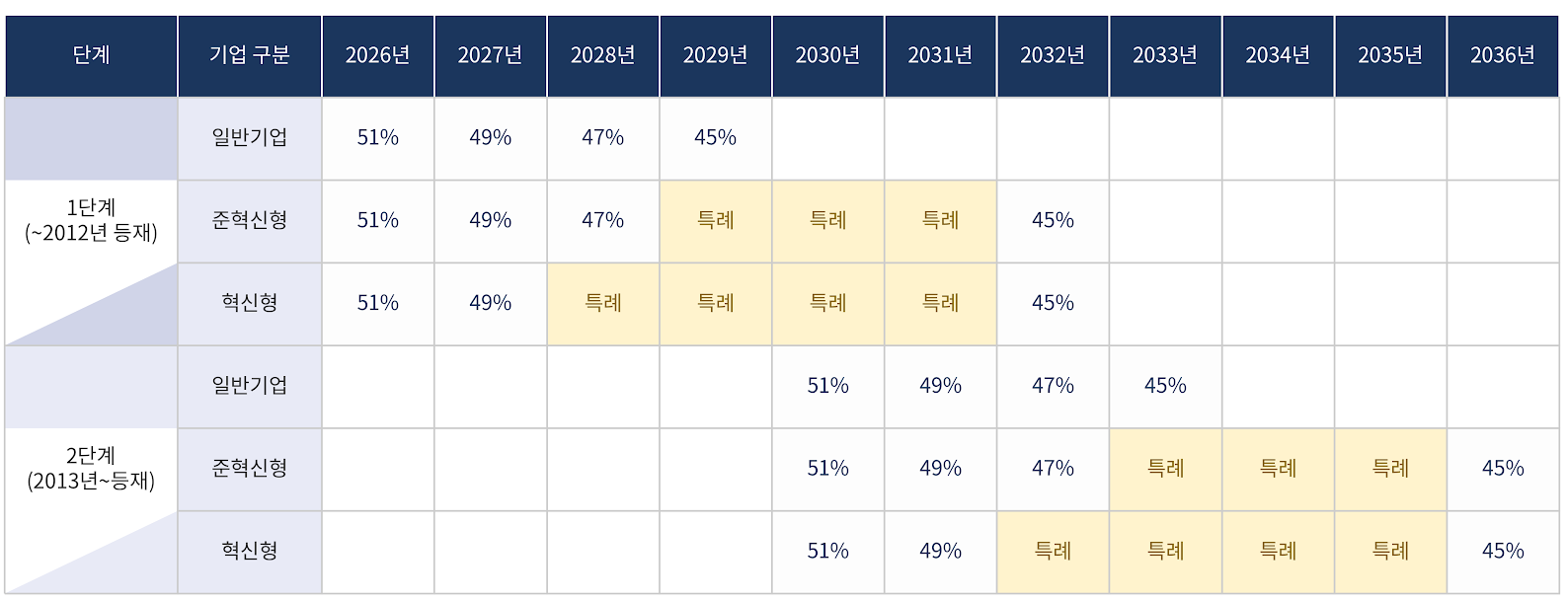
- Generic drug price for non-innovative firms set to 45% by 2029
- by Jung, Heung-Jun Mar 30, 2026 09:12am
- The government will implement price cuts for already-listed drugs, including those of non-innovative pharmaceutical companies, over a period of up to 10 years. With a gradual annual reduction of 2%, the drug price of innovative and quasi-innovative companies is expected to reach a rate of 45% by 2032, while non-innovative companies will reach the same level by 2029.Additionally, for products listed after 2013, gradual reductions will begin in 2030. Non-innovative companies will converge to 45% by 2033, while innovative and quasi-innovative companies will do so by 2036.The aim is to mitigate the shock of a sudden drop in sales for the pharmaceutical industry. Under this structure, if pharmaceutical companies fail to generate new revenue, the scale of their sales decline will gradually increase over the 10-year period.Price reductions for already-listed drugs will be implemented over a maximum of 10 years, including for non-innovative pharmaceutical companies.According to the government’s finalized plan announced on the 26th, the timing of reaching the 45% pricing rate will differ depending on ▲the listing date ▲whether special provisions apply.There are also conditional clauses for drugs with the same active ingredient, those failing to meet requirements, combination drugs, and data-submitted products. This means that even if a product was listed in 2013, it may still be included in the first-stage reduction.First, even general pharmaceutical companies that are not classified as innovative will undergo price cuts over 4 years. Prices will fall to 49% next year, then to 47% in 2028, and to 45% in 2029. Compared to innovative firms, they will reach the 45% level three years earlier.Innovative companies will be given a four-year grace period, and quasi-innovative companies a three-year grace period, reaching 45% by 2032. For innovative firms, the pricing rate will remain at 49% until 2027 and then drop directly to 45% in 2032.Drugs listed from 2013 onward will start at 51% in 2030 and decrease by 2% annually. General pharmaceutical companies will reach 45% in 2033, while innovative and non-innovative companies will similarly receive a 3- or 4-year grace period, with the 45% rate applying in 2036.A key feature is the classification of drugs listed by 2012 and those listed from 2013 into two stages (Stage 1 and Stage 2). However, drugs with the same active ingredient are classified into the same group based on the timing of the first generic entry.For example, even if a drug was listed in 2013, if the first generic for that active ingredient was listed in 2012, it falls under the first stage price reduction. ‘In addition, drugs that do not meet requirements, such as those lacking in-house bioequivalence testing or registered active pharmaceutical ingredients, will be subject to the 45% pricing rate. The same applies to combination drugs and data-submitted drugs that are essentially based on off-patent, generic substances.This means that even if a drug is a combination drug or a data-submission drug, if its active ingredients are equivalent to off-patent generics, it is subject to the 45% adjustment without exception.
- InterView
- [Desk View] Generic bias influencing drug pricing policy
- by Chon, Seung-Hyun Mar 30, 2026 09:12am
- The government decided to lower the price calculation standard for generic drugs from the current 53.55% of the price of the original drug before the patent expiration to 45%. Despite strong opposition from the pharmaceutical industry, the final decision is reportedly similar to the initial draft proposed last year.In November 2023, the Ministry of Health and Welfare (MOHW) reported to the Health Insurance Policy Review Committee a plan to reduce generic pricing to approximately 40%. At that time, the MOHW suggested a plan to adjust drug prices to current levels, ranging from 45-50% to 40%. As products with prices above 45% are classified as targets of adjustment, the government intended that the prices of generic drugs should not exceed that threshold. The original plan was reflected in the final decision.The pharmaceutical industry proposed 48.20% (a 10% reduction from 53.55%) as the figure that they can endure. However, at a Health Insurance Policy Review Committee meeting on the 11th, the MOHW presented a calculation rate in the low-to-mid 40%. Ultimately, while the MOHW secured an alibi of communication over the past five months, the outcome remains largely unchanged from the draft presented nearly half a year ago.While this means that a 45% generic price standard would be reduced by 16.0%, the actual reduction rate grows exponentially when the government's complex price-cutting measures are applied.Under the drug-pricing reform system, the price-cut rate for failing to meet the "highest price requirement" will expand from 15% to 20%. Since July 2020, generic products must meet two conditions to receive the highest price: conducting bioequivalence (BE) studies and using Registered Drug Substances (DMF). For each unmet requirement, the ceiling price drops by a set percentage. Failing both requirements currently results in a 27.75% reduction from the maximum.When applying the new 45% base and the 20% penalty for unmet requirements, generics failing one requirement drop to 36% (a 20.9% drop), and generics failing both requirements drop to 28.8% (a 25.6% drop). By applying a reinforced tiered-pricing system to the drug pricing reform system, entry for late-mover generics would be completely blocked.According to the guidelines following the drug pricing reform in 2020, even if a product meets both standard requirements, if there are already 20 or more identical formulations listed, the 21st product is listed at 85% of the lowest price of the existing formulations or 38.69%, whichever is lower). Currently, the 21st generic drug is set at 32.86%. Compared to the 53.55% maximum, this represents a 38.6% drop for the first product subject to tiered pricing. Prices for the 22nd and 23rd generics drop even further.Under the new reform plan, the 13th generic drug (failing both requirements) would drop to 24.48%. Comparing the same 13th-entry generic, the ratio drops from 53.55% under the old system to less than half under the new reform. By the 13th and 14th entries, prices could drop to as low as KRW 14.98 and KRW 9.20, respectively.Additional mechanisms have been added for price cuts even if a drug was listed at the highest price. To prevent market overheating upon the entry of the first generic, the MOHW intends to apply tiered price cuts to any generic that causes the number of identical formulations to exceed 13. Even if a company is among the first 12 and receives the 45% maximum, if subsequent entries push the total past 13, that product's price will be cut by 15% after one year. This means even the very first generic could see its price drop to 38.25% within 12 months, a 28.58% reduction from the current industry maximum.The pharmaceutical industry argues that the government has meticulously designed to drop drug price over 20% while inhibiting genetic entry of generics.The government's prejudice against generics surfaced amid the drug price reform process. It was based on the prejudice that the increase in generic use could be problematic. Health authorities repeatedly cite rising generic drug expenditures as a threat to the stability of the National Health Insurance (NHI). In 2024, generic drug spending reached KRW 12.44 trillion, a 36.8% increase from KRW 9.09 trillion in 2020. However, the government's argument is that the increased use of generics, which are cheaper than originals, actually saves the NHI budget.Data show that for 14 of 16 dosages among the five most frequently prescribed active ingredients (including atorvastatin, clopidogrel, and rosuvastatin), the weighted-average price is lower than the price of the original drug. This structure demonstrates that as clinical sites prescribe more affordable generics, the overall weighted-average price falls below that of the originals, thereby contributing to fiscal savings. Critics argue the government has focused on statistics on total spending to justify price cuts while ignoring the per-unit savings generics provide.The industry also points out that the government is ignoring existing approvals and regulations.Since July 2020, the number of consigned generics has plummeted because companies must perform their own bioequivalence studies to secure higher prices. Since July 2021, the number of generics that can be approved using a single clinical trial is limited (so called '1+3'). In the past, it was common for dozens of pharmaceutical companies to obtain approval for consigned generics using the same data once a specific company received approval through its own BE testing. However, with the implementation of joint development regulations, 'unlimited replication of generics' is no longer possible.In fact, these regulations have already stifled the entry generics. The number of prescription drug approvals dropped 38% from 4,195 in 2019 to 2,616 in 2020. Last year, only 747 prescription drugs were approved, an 82% drop in six years.The MOHW rationalized the reform by pointing to excessive competition and significant increased number of small-scale firms. The MOHW noted that companies with annual production under KRW 1 billion grew from 54 in 2012 (18.9%). Then, the figure more than doubled to 121 in 2024 (39.3%). However, data details shows that the number of these small-scale firms is declining. The number of firms with an annual finished drug production value of less than KRW 1 billion increased from 51 in 2014 to 124 within just one year. While the growth of these firms slowed starting in 2016, it spiked again to 137 in 2020. Since then, the numbers have declined: down to 133 in 2021 (a decrease of 4 from the previous year) and to 121 in 2024, which is a reduction of 16 companies compared to 4 years prior.The government ignored the impact of the recent regulatory change and overestimated the entry of generics by comparing it with a figure from 10 years prior. Throughout the reform process, the pharmaceutical industry appealed for a policy compromise, citing concerns over reduced R&D investment and job losses.While the government introduced the term "New Innovative Pharmaceutical Company" to offer price incentives for R&D-Intensive firms, the industry remains skeptical of its effectiveness. The industry views the government as having failed to communicate and as having made the system more complex. Ultimately, the industry's distrust of the government has grown.
- Company
- Bispecific antibody Elrexfio lands in Big 5 Hospitals
- by Eo, Yun-Ho Mar 30, 2026 09:12am
- The multiple myeloma drug Elrexfio has secured access to prescribing at major tertiary hospitals in Korea.According to industry sources, Pfizer Korea’s bispecific antibody therapy Elrexfio (elranatamab) has passed the Drug Committees (DC) of Korea’s ‘Big 5’ hospitals, including Samsung Medical Center, Seoul National University Hospital, Seoul St. Mary’s Hospital, Asan Medical Center, and Severance Hospital.However, Elrexfio remains a non-reimbursed drug. Whether it leads to actual prescribing will depend on future reimbursement listing.Although Elrexfio previously passed the Health Insurance Review and Assessment Service (HIRA) Cancer Disease Deliberation Committee review after a second attempt last year, the reimbursement process is currently on hold. Pfizer is expected to pursue reimbursement listing again in the future.Elrexfio, a fourth-line therapy, is an immune cell–engaging treatment composed of two monoclonal antibodies that recognize the target antigen of multiple myeloma and T cells.Elrexfio is a bispecific IgG2 kappa antibody composed of two monoclonal antibodies that respectively recognize BCMA (B-cell maturation antigen), a target antigen of multiple myeloma, and the CD3 antigen. As such, it represents a novel therapy that enables cytotoxic T cells to directly target BCMA-expressing multiple myeloma cells.Multiple myeloma, a cancer of plasma cells in the bone marrow, is a hematologic malignancy that primarily occurs in the elderly. It is a disease where life expectancy can be extended through sustained treatment. While various new drugs are being developed, monoclonal antibodies and bispecific antibody therapies are currently being used in clinical practice.In particular, the bispecific antibody mechanism is considered a safe and effective treatment for relapsed or refractory multiple myeloma, where resistance increases with each treatment cycle, leading to shorter remission periods and fewer available treatment options.Since life expectancy can be extended through continuous treatment, various options must be available for each treatment stage, and securing reimbursement coverage for fourth-line or later treatments is an urgent priority.Currently, bispecific antibody therapies approved in Korea include Elrexfio, as well as Tecvayli (teclistamab) and Talvey (talquetamab), but all remain non-reimbursed. Amid the failed discussions over coverage of a series of bispecific antibody drugs in the early stages, whether any drug will be granted reimbursement and improve patient access is gaining attention.Meanwhile, Elrexfio was designated by the Ministry of Food and Drug Safety as a GIFT item and was approved as a monotherapy for adult patients who have received more than three lines of treatment, including proteasome inhibitors, immunomodulators, and anti-CD38 monoclonal antibodies, in May last year. The US FDA has also designated it as a breakthrough therapy and granted accelerated approval for the drug.Elrexfio’s efficacy was demonstrated through the Phase II MagnetisMM-3 trial, which was conducted on 123 patients who had not received prior BCMA-directed therapy (i.e., BCMA-naïve patients). Results of Cohort A showed that the drug recorded an objective response rate (ORR) of 61.0% and a complete response (CR) of 37.4%.The progression-free survival (PFS) period was 17.2 months, and the overall survival (OS) period was 24.6 months, demonstrating an unprecedented long-term treatment effect. The data demonstrated that Elrexfio provided long-term survival benefits and slowed down disease progression to improve the quality of life of patients who had no other treatment options.

- Policy
- Government busy cutting drug prices without volume control
- by Lee, Jeong-Hwan Mar 30, 2026 09:11am
- Health insurance drug expenditure is calculated by multiplying the reimbursement price by the prescription and utilization volume. No matter how much the government repeatedly lowers drug prices, it is difficult to achieve the policy goal of reducing pharmaceutical expenditure or rationally managing the health insurance budget unless physicians’ prescribing behavior and real-world usage are also controlled.In other words, even if the government lowers the generic pricing rate from 53.55% to 45% (a reduction of about 8.55 percentage points), failure to control prescription and utilization volume could paradoxically lead to higher drug spending than before the price cuts.If not handled carefully, this could lead to a worst-case scenario where the price cut creates an environment in which generics of lower quality than in the past are produced, while at the same time, some companies intensify their efforts to offer illegal rebates to increase prescription volumes in an attempt to mitigate the shock of price cuts.This is why, on the 27th, academia and the pharmaceutical industry urged the Ministry of Health and Welfare not to focus solely on the reform plan for generic drug price cuts, but to establish mechanisms to reasonably control prescription volumes.Academia “Government must lead generic competition through policies such as mandatory lowest-price substitution”Academia argues that attempting to reduce health insurance expenditure through price cuts alone without addressing volume control is fundamentally misguided.In particular, the prevailing view among academics is that while the Ministry’s efforts are needed to create an environment where pharmaceutical companies can compete to dominate the market through “low-cost generic” strategies, its repeated exclusion of this aspect is ultimately reinforcing an industry structure that leaves companies with no choice but to generate sales through illegal rebate competition.Criticism has been raised that the Ministry has virtually no plans for policies designed to enable low-cost generics to gain a competitive edge in the market, such as promoting the substitution of the lowest-priced generics, establishing a competitive landscape for generics based on international nonproprietary name prescriptions, or shifting the environment for generic selection through a Korean-style reference pricing system.Health economists emphasize that once bioequivalence is established, generics should be allowed to compete solely on price.Professor Hye-young Kwon of Mokwon University stated, “The conflict between the industry and the government over the generic drug pricing rate is meaningless. If drug prices fall, companies are more likely to increase prescription volume through rebates, which may maintain or even increase overall drug spending. Ultimately, cheaper generics must be prescribed more and sold more in order to achieve both health insurance savings and pharmaceutical industry development.”Professor Kwon added, "The Ministry of Health and Welfare must devise administrative measures to create a competitive market where pharmaceutical companies that lower prices the most for each ingredient dominate the market. A pharmaceutical company that wins on price in the domestic market has a good chance of succeeding in the global market as well. The government must work with doctors and patients to devise policies such as mandating the substitution of the lowest-priced generic, using the cheapest drug as a reference price, and providing incentives when that drug is used, or imposing disincentives when more expensive generics or originals are used.”She emphasized, “If we establish a generic price competition policy that goes beyond the government and pharmaceutical companies to include doctors, pharmacists, and patients, pharmacists and patients will be able to intervene in the decision-making process to ensure cheaper drugs are used, even if a doctor unilaterally prescribes a specific drug This would allow rationalization of health insurance finances without excluding physicians’ clinical judgment. The government needs to make a decisive move and establish the necessary framework.”Professor Seung-jin Bae of Ewha Womans University College of Pharmacy also believes that the Ministry of Health and Welfare has not given sufficient consideration to usage policies beyond generic price adjustment policies. In particular, the professor stated that administrative measures by the Ministry are needed to address the current reality where doctors are bound to brand-name prescriptions despite the government recognizing generics as therapeutically equivalent.Specifically, Bae suggested activating lowest-price substitution or implementing INN prescribing to enable genuine price competition among generics.Bae said, “We need policies that allow cheaper generics to expand market share. The issue of brand-name prescribing must be addressed first. It makes no sense for the government to acknowledge bioequivalence and still tolerate brand-name prescribing practices.”Bae emphasized, “We should not view this (the development of health insurance cost-saving policies) as a conflict between professions. Instead, we must consider measures such as substituting the lowest-priced generic to allow the market for chemical generics to be organized through price competition. Korea’s health insurance resources are not unlimited. In the case of biosimilars, there may be differing opinions regarding INN prescribing or substitution. However, for chemical drugs, we must take a more proactive approach from the perspective of health insurance finances.”The domestic pharmaceutical industry also maintains that, alongside price cuts, addressing physician prescribing behavior and patient overuse is essential to achieving effective cost savings. If the government pays attention to volume control, it could move away from its current administrative pattern of repeatedly relying on price cuts.An official from a leading domestic pharmaceutical company pointed out, “The government establishes policies claiming it will reduce drug costs, maintain generic drug quality, ensure a stable supply of medicines, develop innovative new drugs, and foster robust pharmaceutical companies, all within limited health insurance funds, but the conclusion always boils down to drug price cuts. It is time to consider the root causes of why we face criticism that medical institutions prescribe more drugs than necessary and why pharmaceutical companies are forced to boost sales through rebate competition.”“If we merely suppress drug prices while neglecting the increase in usage, we cannot achieve fiscal savings. If we only cut drug prices, companies will find it difficult to avoid decisions such as increasing prescriptions for their own drugs through aggressive marketing using CSOs, or producing and supplying low-quality generics by cutting costs and reducing employment.”Calls grow for governance involving government, industry, and academia in drug pricing policyWhenever drug price reduction reform plans are discussed, some in the pharmaceutical industry offer a self-deprecating assessment that “pharmaceutical companies are always in the position of the weakest party.”This reflects the reality that pharmaceutical companies find it difficult to openly and freely express their opinions or arguments, as they must constantly gauge the reactions of both the Ministry of Health and Welfare, which seeks to lower drug prices, and doctors, who hold the power to prescribe medications.Accordingly, the pharmaceutical industry is requesting that, starting with this drug pricing system reform proposal, a governance structure be established where government agencies, the pharmaceutical industry, and academia can discuss and design drug pricing policies together.The intent is to ensure that the pharmaceutical industry is granted the authority to participate in the decision-making structure for establishing drug pricing policies, including drug price reductions and revisions to post-approval drug price management systems, both administratively and legally.In the recent reform process, since the Ministry’s proposal was announced on November 28 last year, the pharmaceutical industry has been limited to merely proposing amendments and improvements to the Ministry’s draft, rather than actively voicing its own opinions, from the time the Ministry’s revised proposal was submitted to the subcommittee of the Health Insurance Policy Deliberation Committee on March 11 of this year until its final approval at the plenary session on the 26th.In particular, since the Ministry’s proposal was not made public until just before it was reported to the HIPDC, submitted to the subcommittee, and finally approved at the plenary session, pharmaceutical companies’ drug pricing officials had no choice but to work tirelessly trying to interpret the government’s intentions.In contrast, advanced countries overseas guarantee the pharmaceutical industry’s right to submit opinions on the direction, implementation timeline, and detailed regulations of drug pricing system reform proposals through mandatory agreements or legal commitments at the executive and industry levels.France operates under a framework agreement between CEPS and the pharmaceutical industry association, while the UK mandates stakeholder participation in pricing and reimbursement reforms through NHS England and the Department of Health.Japan also determines pricing through the Central Social Insurance Medical Council under the Ministry of Health, Labour and Welfare.An official from a mid-sized pharmaceutical company stated, “This drug price reform should serve as an opportunity to prepare various measures for establishing public-private consultative governance and to submit industry-level opinions to the government and the National Assembly. Even if we cannot exert a substantial influence on the establishment of the system, wouldn’t this create a forum to publicly convey the pharmaceutical industry’s well-founded opinions to the government, academia, and the public?”Academics have also suggested that improving the operational standards of the Health Insurance Policy Deliberation Committee (HIPDC), which is already run by the government, could ensure transparency in the establishment of the drug pricing system and the national health insurance system.Professor Bae said, “Regarding the governance of the drug pricing system and national health insurance policies, there is a need to ensure transparency by disclosing meeting minutes and other details discussed within the HIPDC. National health insurance finances and policies are funded using premiums paid by the public. Since the system is funded by public contributions, citizens have the right to know what is discussed during the Health Insurance Policy Deliberation Committee’s proceedings.”He added, “Ultimately, for everyone to accept the health insurance policies decided by the government, we must improve the process so that it is clear who expressed what opinions at the committee, rather than suddenly bringing items to a vote as is currently done. Creating additional consultative bodies outside the Health Insurance Policy Deliberation Committee framework risks creating unnecessary layers of bureaucracy.”