- LOGIN
- MemberShip
- 2026-06-14 08:30:06

- Company
- MedTech–Pharma alliances intensify as remote monitoring turns profitable
- by Hwang, byoung woo Mar 19, 2026 12:39pm
- As the remote patient monitoring (RPM) market enters a profit-generating phase, competition in the form of alliances between medical device companies and pharmaceutical firms is intensifying.Currently, the domestic RPM market is developing within a framework of alliances between medical device companies and pharmaceutical firms. Seers Technology is partnering with Daewoong Pharmaceutical, Mezoo with Dong-A ST, Huinno with Yuhan Corp, and Wellysis with Samjin Pharmaceutical, forming a competitive landscape among these alliances.Seers Technology is rapidly building references by targeting hospitals with its ward monitoring platform thynC. Mezzue, which is set to list on KOSDAQ in March, is targeting the ward monitoring market with its mobile patient monitoring platform, HiCardi. Wellysis is expanding through wearable ECG devices, while Huinno is growing its business around ECG-based digital healthcare solutions.This surge in collaboration is interpreted as a sign that the RPM market has entered a phase where it is generating revenue. Pharmaceutical companies can expand patient management services based on therapeutic expertise and sales networks, while digital healthcare firms can strengthen data acquisition and platform competitiveness. These aligned interests are accelerating collaboration.RPM market expands beyond ECG to patient management platformsRPM is a digital healthcare technology that continuously measures and manages patients’ vital signs outside hospitals or within general wards.While the initial market was centered on wearable ECG-based arrhythmia detection technology, it has recently been rapidly evolving into a platform format that simultaneously analyzes various vital signs such as heart rate, respiration, body temperature, and physical activity levels.According to market research firm Markets and Markets, the global RPM market is expected to grow from USD 27.7 billion (KRW 41 trillion) in 2024 to USD 56.9 billion (KRW 85 trillion) by 2030, with an estimated annual growth rate of approximately 12–13%.RPM market prospect: Rise from USD 27.7 billion (2024) to USD 56.9 billion (2030) In fact, as hospitals increasingly adopt ward-based monitoring systems, related companies are seeing rapid revenue growth.Industry observers assess that the RPM market is now transitioning from the technology validation phase to the actual commercialization phase.An industry source stated, “Pharmaceutical companies are expanding the market by applying specialized strategies aligned with their therapeutic areas and sales environments. Even those not currently collaborating with medical device companies are closely watching as successful cases emerge.”Hospitals adopt multiple platforms …market expansion phaseHowever, many in the industry view the competition in the RPM market as being closer to a phase of market expansion rather than a zero-sum game for the time being.In fact, some hospitals are adopting multiple RPM platforms simultaneously.An industry insider noted, “While some hospitals adopt only a single solution, others adopt two platforms simultaneously after proof of concept (PoC) has been validated. Since the RPM market is still in its early stages, the structure allows multiple companies to grow together.”Similar to how markets expand when new mechanisms of action enter therapeutic areas, insiders believe the key factor for the RPM market is in increasing the number of hospital beds adopting the technology.As a result, analysts expect the RPM market to grow rapidly over the next 3–5 years, with multiple players expanding simultaneously.Competition in the RPM market is expected to intensify further following upcoming initial public offerings (IPOs) and global expansion.For example, Daewoong Pharmaceutical is pursuing a platform expansion strategy involving multiple partners in addition to Seers Technology, such as Skylabs, iKooB, and Puzzle AI, suggesting that competition may evolve beyond simple partnerships between medical device companies and pharmaceutical firms, into broader digital health alliances.Ultimately, the RPM market may evolve beyond mere collaboration between medical device companies and pharmaceutical firms into a data-driven patient management platform industry.An industry insider stated, “The RPM market is now moving beyond technological competition into a phase of platform competition. How companies build service models based on patient data will determine their future competitiveness.”

- Company
- Se Eun Hwang appointed as the new general manager of CSL Korea.
- by Son, Hyung Min Mar 19, 2026 12:39pm
- Se Eun Hwang, new General Manager of CSL KoreaCSL announced the appointment of Se Eun Hwang, former general manager of Biogen Korea, as the new General Manager of its Korean subsidiary.Effective the 17th, Hwang will oversee overall operations of CSL Korea, leading domestic business strategy, strengthening organizational capabilities, and driving patient-centric corporate operations.Hwang has extensive leadership experience across Korea’s pharmaceutical and biotech industry, covering commercial strategy, market access, and medical affairs.She has successfully driven business growth by building competitive organizations and successfully launching products in Korea’s complex regulatory environment.Before joining CSL, Hwang served as general manager of Biogen Korea, where she established the local organization and led its expansion. During her tenure, she successfully developed reimbursement strategies and achieved market entry for rare disease therapies.Previously, she served as Head of a Rare Disease Franchise at Handok and as Head of Marketing at Abbott Korea, and she also held key positions at JW Pharmaceutical and Hanil Pharmaceutical, gaining experience across the entire pharmaceutical industry. Hwang is a pharmacist by training. She earned an MBA from Sogang University and subsequently obtained a Ph.D. in Clinical Pharmacy from Chung-Ang University, specializing in Social and Health Pharmacy.The newly appointed general manager said, “I am both delighted and feel a strong sense of responsibility taking on this important role, which will allow me to contribute to the lives of patients with rare and severe diseases in Korea through CSL’s innovative portfolio. We will prioritize patient-centric values, maximize organizational capabilities, and work closely with partners to drive new growth at CSL Korea.”Through the leadership appointment, CSL plans to further strengthen its business foundation in Korea and continue efforts to improve the quality of life for patients with rare and severe diseases.

- Company
- Dayvigo targets Zolpidem dominance…Korean commercial launch imminent
- by Eo, Yun-Ho Mar 19, 2026 12:39pm
- An insomnia treatment with a new mechanism of action, ‘Dayvigo,’ is soon to be commercialized in Korea.According to industry sources, Eisai Korea is currently undergoing the approval process with the Ministry of Food and Drug Safety (MFDS) for Dayvigo (lemborexant), a dual orexin receptor antagonist (DORA). Formal approval is anticipated around mid-year.Mechanistically, Dayvigo is classified as an orexin/hypocretin receptor antagonist. It works by inhibiting orexin, which promotes sleep. It is important to note that orexin itself is a neuropeptide in the brain that promotes wakefulness.The drug demonstrated efficacy through two Phase III clinical trials, including the SUNRISE I study.The trials involved 1,006 adult patients from 67 medical institutions in the U.S. and Europe, who were divided into 4 treatment groups. These included 266 patients in the Davigo 5 mg treatment group, 269 in the Davigo 10 mg treatment group, 263 in the zolpidem CR Tab (6.25 mg) treatment group, and 208 in the placebo group. The average age of the patients participating in the study was 63, and 86% were female.Key results showed that most patients in the Dayvigo groups fell asleep within 20 minutes, demonstrating improved sleep onset. In addition, nighttime sleep maintenance time increased by more than 60 minutes compared to baseline.Furthermore, both the low-dose and high-dose groups demonstrated superiority over the placebo group in terms of sleep onset and sleep maintenance. These improvements were particularly pronounced in the Dayvigo 10mg treatment group.Dayvigo received FDA approval in 2019 and is currently prescribed in markets including Europe and Japan.
- Company
- Jeil's subsidiary, Onconic licensing transactions total KRW 10B
- by Chon, Seung-Hyun Mar 19, 2026 12:39pm
- Jeil Pharmaceutical has reached the cumulative technology licensing fees revenue of KRW 10 billion from its R&D subsidiary, Onconic Therapeutics. This steady influx of technology licensing fee stems from the completion of domestic clinical trials, the approval of the new drug Jaqbo, and its successful entry into the Chinese market. Additionally, Jeil Pharmaceutical's revenue surged to KRW 67.1 billion last year as it ramped up sales of Jaqbo.According to the Financial Supervisory Service (FSS) on the 18th, Jeil Pharmaceutical received a total of KRW 1.8 billion in technology licensing fees from Onconic Therapeutics last year. Specifically, the company received KRW 600 million in February, followed by KRW 300 million and KRW 900 million in May and October, respectively.Yearly Cumulative Technology Licensing Fees of Jeil Pharmaceutical's subsidiary Onconic Therapeutics (unit: KRW 1 million, source: Financial Supervisory Service)These payments stem from the settlement of technology export milestones for Jaqbo, which Jeil Pharmaceutical originally out-licensed to Onconic Therapeutics. In March 2023, Onconic Therapeutics signed a technology export deal with the Chinese firm Livzon Pharmaceutical Group for Jaqbo, valued at up to $127.5 million. Under the terms, Onconic received a non-refundable upfront payment of $150 million and is expected to receive up to $112.5 million in milestone payments based on development, regulatory approval, and commercialization stages.In January of last year, Onconic Therapeutics received a $3 million milestone from Livzon following the first patient dosing in China's Phase 3 clinical trial. In March, it billed an additional $1.5 million after completing the transfer of production technology to Livzon.By August, Onconic billed and received a $5 million development milestone within 30 business days. This was achieved by the successful conclusion of Phase 3 trials in China and the submission of a New Drug Application (NDA) to the National Medical Products Administration (NMPA). A portion of these fees received from Livzon is redistributed to Jeil Pharmaceutical as milestones.Jaqbo is a P-CAB (Potassium-Competitive Acid Blocker) drug, approved in April 2024 as the 37th Korea-developed new drug. P-CAB anti-ulcer agents work by competitively binding with potassium ions to the proton pump, the final stage of acid secretion in gastric parietal cells, thereby inhibiting gastric acid secretion.Jeil Pharmaceutical's revenue history from Jaqbo includes two non-refundable payments of KRW 150 million each in December 2020 and May 2021. In 2022, it received KRW 1 billion in February and another KRW 1 billion in December as Phase 3 clinical milestones. In April 2023, the company received KRW 2.7 billion as a settlement from Onconic’s upfront payment from Livzon (representing 13.5% of the $15 million). In 2024, an additional KRW 600 million was paid over four installments. As of last year, Jeil's cumulative technology licensing fees for Jaqbo totaled KRW 7.58 billion.Jeil Pharmaceutical has also consistently generated revenue from an oncology drug out-licensed to Onconic. For the dual-target anticancer agent JPI-547, the company has secured a total of KRW 2.5 billion. This includes upfront payments of KRW 750 million each in December 2020 and May 2021, plus a KRW 1 billion development milestone received in December 2022. The total amount Jeil Pharmaceutical has collected from Onconic Therapeutics for both Jaqbo and JPI-547 amounted to KRW 10.08 billion.Through Jaqbo domestic sales, Jeil Pharmaceutical achieved expanded sales effects. Jaqbo entered the prescription market in last October after receiving National Health Insurance coverage. Jeil Pharmaceutical and Dong-A ST collaborate on leading marketing and sales. Jaqbo's revenue for Jeil expanded from KRW 8.3 billion in 2024 to KRW 67.1 billion last year.
- Policy
- "MOHW will design the drug pricing policy considering NHI cost"
- by Lee, Jeong-Hwan Mar 19, 2026 12:38pm
- Lee Hyung-hoon, Second Vice Minister of Health and Welfare"We recognize that the pharmaceutical industry has varying opinions regarding government policy. However, the government designs policy by looking at where the public's expectations lie and what their interests are. The necessity of fostering the pharmaceutical industry and the fact that domestic generic drug prices are high are areas where society relatively shares a consensus, and social agreement is required. We are not setting a single specific drug pricing reform plan and trying to force it through. We will repeatedly collect and discuss opinions from the pharmaceutical industry. Since policy can change at any time, there is room to modify the reform plan."Lee Hyung-hoon, Second Vice Minister of Health and Welfare (60, Yonsei University), has drawn significant attention by highlighting the difficulties of reconciling diverse interests in a single policy for drug pricing reform, which primarily focuses on lowering the prices of previously-listed generic drugs.Lee stated that, given South Korea's adoption and operation of a single-payer National Health Insurance system, writing a reform plan that simultaneously achieves the goals of fostering the pharmaceutical industry and ensuring efficient, rational national pharmaceutical expenditures requires considering the interests of both the industry and the public as beneficiaries of health insurance.Meeting with the Korea Special Press Association on the 18th, Vice Minister Lee promised, "Rather than taking the stance of a policy authority, the Ministry of Health and Welfare designs policy by examining public interests. However, we have not finished setting a single drug pricing reform plan. Policy is open and can continue to change. We will communicate with the pharmaceutical industry."This was the Vice Minister's response when asked how the Ministry intends to resolve the industry's backlash against the drug pricing system reform plan announced on November 28 last year.Lee stated that, while agreeing on the necessity of fostering the South Korean pharmaceutical and biotech industry on a global scale to strengthen national competitiveness, the final reform plan would be finalized after collecting opposing opinions from the industry on uniform price cuts.The Ministry intends to listen to the industry's voice and to prepare a revised proposal until the Health Insurance Policy Deliberation Committee deliberates on the reform plan on the 26th.Since releasing the initial draft of the reform plan on November 28 last year, the Ministry has presented a revised draft to the industry on March 11 at the subcommittee. However, the domestic pharmaceutical industry continues to criticize the process as 'closed administration,' citing the failure to specify clear generic price calculation rates, mentioning only a range of 'low-to-mid 40%,' the lack of specific timing and methods for price cuts per item, and the absence of detailed criteria and periods for preferential drug pricing.Lee repeatedly stated that he would gather industry opinions before the committee's final decision. He emphasized that, because pharmaceutical expenditures are funded through the health insurance budget, the government views the drug pricing system considering the efficiency of public insurance premiums."South Korea operates the drug pricing system based on the health insurance finances funded by the mandatory premiums paid by citizens," Lee stated. "Because medical fees and drug prices for both generics and new drugs are operated through health insurance finances, we value the National Health Insurance cost, which represents public expectations."Lee also noted that he recognizes the domestic pharmaceutical industry as a partner in South Korea's efforts to advance to one of the top five global pharmaceutical and biopharmaceutical powers. This is interpreted as a necessity for the administration to communicate and make adjustments to the aspects the industry hopes will be reflected in the reform plan.Lee stated, "Since the Ministry of Health and Welfare also aims to leap into a top-five pharmaceutical and bio-powerhouse as a national task, the pharmaceutical industry is an important partner," Lee explained. "We are discussing the reform plan with the industry. There is a backlash because the industry has more favorable, explicit expectations, but we will continue to communicate. There is room for modification within the system."Lee added, "There may be differing opinions because the industry has its own perspective and expected levels. While the Ministry has its position as a policy authority, we work by looking at where the public views, perspective, and interests lie."Lee stated, "We are open to differing opinions on Ministry policy or points raised by the media. We are not setting one thing and trying to force it through," adding, "Policy can continue to change."Lee concluded by saying, "The need to foster the pharmaceutical industry and the fact that generic prices are high are areas where there is a relatively social consensus. We have carefully considered the drug pricing policy. We are listening to the pharmaceutical industry's opinions," adding, "We fully recognize, acknowledge, and respect the necessity for innovative pharmaceutical companies and the domestic industry to possess national competitiveness and move toward becoming a bio-power. We support and share with the industry's aspiration to expand to a larger global stage."
- Company
- Multi-indication anti-cancer drugs, 'Tevimbra' offers alternative
- by Eo, Yun-Ho Mar 18, 2026 09:12am
- Product photo of 'Tevimbra (tislelizumab)' As advanced oncology treatments surge, discussions are in full swing to bridge the gap between the financial burden and improved patient access.The government recently presented the enhancement of coverage for rare cancers as a core task through the 5th Comprehensive Cancer Control Plan (2026–2030), and the need to re-examine the overall financial structure of oncology reimbursement is gaining attention.Related to this, the Korean Society of Medical Oncology has also emphasized the need to expand the scope of reimbursement for rare cancer treatments, calling for a balance between coverage expansion and fiscal sustainability.The background to these policy discussions is the rapid expansion of approved uses for multi-indication oncology drugs, such as immune checkpoint inhibitors, antibody-drug conjugates, and bispecific antibodies, which could accelerate the pace of expenditure growth.The recent regulatory and expanded reimbursement of the immunotherapy 'Tevimbra (tislelizumab)' is garnering significant attention. It is being interpreted as a case study demonstrating how realistic alternatives can be implemented amidst spending on multi-indication oncology drugs is skyrocketing.Tevimbra entered the Korean market in April last year, becoming the first immunotherapy to successfully secure reimbursement for the first-line treatment of esophageal cancer in combination with chemotherapy. Within just two months, it expanded its approved indications to a total of five, including esophageal cancer, gastric cancer, and first- and second-line treatment of non-small cell lung cancer.In December of the same year, an unusual record for Tevimbra was set when all five of these indications passed the Cancer Drug Review Committee in a single session. After that, it secured additional indications in perioperative adjuvant therapy for non-small cell lung cancer, extensive-stage small-cell lung cancer, and nasopharyngeal cancer. This drug's scope was expanded from major to rare cancers with limited existing treatment options in a short period. However, the expanded reimbursement for major indications remains ongoing.Despite concerns in the medical community that regulatory and reimbursement hurdles are rising, Tevimbra's rapid achievement is raising expectations, largely due to a strategy that combines proven clinical equivalence with a rational pricing model. By demonstrating clinical utility equivalent to first-in-class agents while offering a pricing structure that reduces the financial burden relative to competitors, the drug has navigated the approval and reimbursement processes swiftly. Experts evaluate Tevimbra as a symbolic case that tests the consistency of the reimbursement review process.Both clinicians and National Health Insurance subscribers are also welcoming Tevimbra as an expansion of treatment options. Clinicians appreciate the increased choices within the same efficacy profile, allowing for prescriptions tailored to patient characteristics. At the same time, payers view the situation positively as price competition between drugs with equivalent clinical effects can lead to significant cost savings.Providing options for rare cancers like nasopharyngeal cancer, or for areas like perioperative non-small cell lung cancer and esophageal cancer where reimbursed treatment options were previously lacking, aligns with the government's goal of expanding reimbursement coverage.In a situation where the reimbursement system is becoming restricted due to spending being concentrated on specific immunotherapies, a drug that successfully enters the reimbursement list by presenting a new price structure based on clinical similarity is expected to set a favorable precedent.Professor Ji-Youn Han of the Hematology-Oncology Department at the National Cancer Center said, "Tevimbra demonstrated an efficacy and safety profile equivalent to that of existing immunotherapies across various studies. In certain patient subgroups, it has shown clear relative advantages." Professor Han added, "As clinical evidence for Tevimbra grows, it is an option that can broaden the scope of treatment selection and improve clinical benefits and access. We hope for rapid reimbursement expansion so that patients can receive benefits."
- Policy
- MOHW drug pricing reform to likely bypasses NA briefing
- by Lee, Jeong-Hwan Mar 18, 2026 09:11am
- Joomin Park, chair of NA Health and Welfare CommitteeThere is a growing likelihood that the Ministry of Health and Welfare’s drug pricing system reform plan, the first in 14 years, will be finalized and implemented without a formal briefing to the National Assembly.Although Joomin Park, Chair of the National Assembly Health and Welfare Committee from the Democratic Party of Korea, instructed Health and Welfare Minister Jung Eun-kyung to provide a separate briefing on the reform plan, it has been confirmed that the ruling and opposition floor leaders on the committee are not actively coordinating a schedule for a plenary session.On the 17th, an official from an opposition lawmaker’s office on the National Assembly’s Health and Welfare Committee explained, “We have heard absolutely nothing regarding a plenary session for a separate briefing on the drug pricing reform plan.”If this explanation is correct, it means that despite Chair Park’s remarks on the need for a separate briefing on the drug pricing system, negotiations between Democratic Party floor leader Sujin Lee and People Power Party floor leader Miae Kim have come to a complete standstill. If an additional plenary session for the briefing is not held, the Ministry of Health and Welfare will proceed with the drug pricing reform plan, which has faced backlash from the pharmaceutical industry, without reporting to the National Assembly.The need for an additional National Assembly briefing on the drug pricing reform plan first arose during the plenary session held on the 10th for the annual policy briefing by relevant government ministries, when Rep. Sun-min Kim of the Rebuilding Korea Party raised the need through a procedural intervention.Rep. Kim criticized the ministry for attempting to push forward rapidly with the drug pricing reform—centered on across-the-board generic price cuts—despite opposition from the pharmaceutical industry, while including no update at all on the reform plan among the key issues in its official policy briefing.At the time, Committee Chair Park agreed with Kim and told Minister Eun-Kyoung Jeong, “Because this is a very important matter, it would be a good idea to provide an additional briefing at a plenary session once the procedures related to the reform plan are completed.”Nevertheless, the reason why the convening of the Welfare Committee’s plenary session remains a distant prospect appears to be rooted in the Ministry of Health and Welfare’s passive attitude and the suspension of consultations between the ruling and opposition parties.The Ministry of Health and Welfare has no incentive to take the lead in convening a plenary session at the administrative level, free from the burden of a one-point briefing, as the reform plan to lower the generic drug pricing rate from 53.55% to the 40% range has met with opposition from the pharmaceutical industry.As for the ruling party, given the ongoing fierce criticism from the opposition regarding the national vaccination campaign for COVID-19 vaccines found to contain foreign substances, which proceeded without proper procedure, the party has no reason to hold an additional plenary session that would expose it to further attacks from the opposition.In fact, People Power Party lawmakers on the Legislation and Judiciary Committee have decided to file a complaint accusing Minister Eun-kyoung Jeong, who served as Commissioner of the Korea Disease Control and Prevention Agency during the COVID-19 pandemic, of alleged dereliction of duty over the administration of 14.2 million doses of contaminated vaccines.Time constraints also make an additional plenary session difficult. The ministry plans to finalize the details and implementation schedule of the drug pricing reform at the HIPDC meeting on the 26th, and physically, it may be impossible to hold an additional National Assembly briefing before then.If the additional plenary session of the Health and Welfare Committee fails to materialize, the ministry is expected to have the reform plan approved at the HIPDC without first reporting it to the National Assembly.The Ministry of Health and Welfare plans to finalize the specific details and implementation date of the reform plan at the full HIPDC meeting on the 26th, following a subcommittee meeting on the 11th focused solely on the drug pricing reform plan and another subcommittee meeting on the 18th.An official from an opposition party lawmaker’s office on the Health and Welfare Committee said, “It is uncertain whether a plenary session for an additional briefing on the drug pricing reform plan will be held. Time is tight physically, and I understand that both the ruling party and the ministry are passive about holding one. The conflict between the ruling and opposition parties over the contaminated vaccine issue and calls for Minister Jeong’s accountability are also affecting the failure to agree on the briefing for the drug pricing reform plan.”The official continued, “In a situation where the opposition is demanding a hearing on contaminated vaccines and the indictment of Minister Jeong, an additional briefing on the drug pricing reform plan, which is already drawing strong resistance from the pharmaceutical industry, would naturally be burdensome for the ruling party. Although the pricing reform is an important policy that will shape the future of the pharmaceutical industry, and although Chair Park has requested a report on it, the government and ruling party seem to lack the will to proceed.”An official from the office of Democratic Party floor leader Sujin Lee stated, “Given the timing, it seems difficult to hold an additional plenary session on the drug pricing system, but we plan to hear the opinions of the opposition party’s floor leadership. If an additional briefing proves difficult, we will also consider a plan for ruling party lawmakers to receive individual briefings on the reform plan from the Ministry of Health and Welfare.”
- Company
- 'Nubeqa' reimb progress…changes to prostate cancer trt strategies
- by Son, Hyung Min Mar 18, 2026 09:11am
- Prostate cancer treatment 'Nubeqa' The prostate cancer treatment 'Nubeqa' is under reviews for final insurance reimbursement hurdle.As the competing drug 'Erleada' faced setbacks in price negotiations and the patent expiration of Xtandi approaches, a total shift in the competitive landscape among Androgen Receptor Pathway Inhibitors (ARPI) is expected.According to industry sources, Bayer Korea is set to enter price negotiations with the National Health Insurance Service for Nubeqa (darolutamide). Nubeqa passed the Drug Benefit Evaluation Committee earlier this month, advancing to the final stage of coverage.The indications that passed the reimbursement evaluation include ▲high-risk non-metastatic castration-resistant prostate cancer ▲hormone-sensitive metastatic prostate cancer (mHSPC) in combination with androgen deprivation therapy (ADT) ▲mHSPC in combination with docetaxel and ADT.Among these, the hormone-sensitive combination therapies were approved as suitable for reimbursement on the condition that a price below the evaluated amount be accepted.Once reimbursement discussions are finalized, Nubeqa will be available for practical clinical use in both triplet and doublet therapies. The strength of this drug lies in its differentiated treatment strategies, depending on whether docetaxel is co-administered, allowing for customized treatment tailored to the patient's condition.Doctors evaluate that this will expand treatment options, as triplet therapy (Nubeqa+ADT+docetaxel) can be used for patients who require aggressive early treatment. In contrast, doublet therapy (Nubeqa+ADT) can be considered for elderly patients or those with high chemotherapy burdens due to comorbidities.Clinical evidence for Nubeqa continues to accumulate, with the ARANOTE study targeting mHSPC patients showing that the Nubeqa + ADT doublet therapy significantly improved outcomes, reducing the risk of radiological progression or death by 46% compared to placebo.It also delayed the time to progression and PSA progression, showing consistent results across secondary endpoints. A post-monitoring analysis confirmed delay in time to deterioration of quality-of-life and pain progression.Furthermore, the ARASENS study showed that the triplet combination of Nubeqa + ADT + docetaxel significantly improved overall survival, reducing the risk of death by 32.5%.Competition among ARPIs intensifies…the market is closely watching variables such as reimbursement and patentsCurrently, the prostate cancer treatment landscape is centered on androgen receptor inhibitors.Major treatment options include Janssen's 'Zytiga (abiraterone),' 'Erleada (apalutamide),' and 'Xtandi (enzalutamide)' serving as major options alongside Nubeqa.However, several variables surround the market environment. Competing drug Erleada reportedly failed to reach an agreement during recent price negotiations with the National Health Insurance Service on expanding reimbursement for its high-risk non-metastatic indication.Prostate cancer treatment 'Xtandi'Analysis suggests that the adjustment of the Risk-Sharing Agreements (RSA) rate following the expansion of the target population is a key point of contention.Furthermore, the patent for Xtandi, which has led the market, is set to expire in major countries, starting with the U.S., in 2027. As Xtandi is a blockbuster with annual sales of approximately $6 billion, the possibility of structural market changes due to generic entry is being discussed in the mid- to long-term.Ultimately, Nubeqa's reimbursement progress, beyond a new listing, is evaluated as a variable that will influence the overall competitive landscape.As treatment options with clinical use as both doublet and triplet combination strategies are made available, depending on reimbursement status, the paradigm of prostate cancer treatment in Korea is increasingly likely to be restructured toward more patient-centered care.
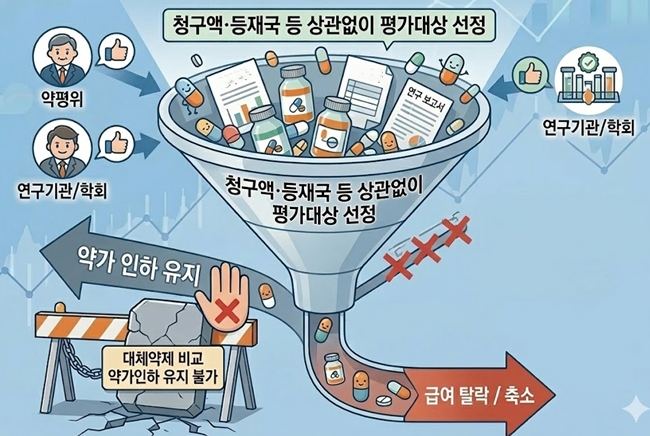
- Policy
- Scope expanded, outcomes tightened for reimbursement reassessment in Korea
- by Jung, Heung-Jun Mar 18, 2026 09:11am
- The government is moving to specify implementation criteria as part of its efforts to reform the reimbursement adequacy reassessment system. It appears that ingredients for which the Drug Reimbursement Evaluation Committee (DREC), research institutes, academic societies, and others raise the need for reassessment will undergo reevaluation regardless of claim volume or listing-country criteria.In addition, the option of maintaining reimbursement through price cuts by comparing a product with its alternatives is expected to disappear going forward.The government is specifying the implementation criteria for reimbursement adequacy reassessment. AI-generated image.According to industry sources on the 17th, the government is specifying the implementation requirements for reimbursement reevaluation reassessments ahead of deliberation by the Health Insurance Policy Deliberation Committee (HIPDC) later this month.Until now, reassessments have been conducted on ingredients among older listed drugs that met the claim volume and countries of listing criteria.The reform plan announced last November signaled a shift away from criteria based on claim volume and listed countries, instead focusing on: ▲ ingredients for which national health authorities in A8 countries have initiated clinical or reimbursement adequacy reassessment; ▲ cases where data or clinical evidence contradicting previously reported efficacy has been published; and ▲ drugs for which the need for reassessment has been recommended by academic societies or experts.Recently, ingredients deemed necessary by DREC through monitoring, as well as those evaluated by research institutions or public agencies, have been subject to additional review.This means the committee will be able to proactively examine and designate a broader range of candidates for reassessment. As a result, concerns are emerging within the industry that the number of drugs subject to reassessment may increase.A pharmaceutical industry official said, “There had already been a tendency to broaden the criteria and expand the scope of targets. The intent is to cut drug spending by strengthening reassessment. Since the idea is that reassessment will proceed whenever there is a perceived need, I believe the number of targets will likely increase.”In particular, because the results of reassessment are being simplified into either delisting or positive listing, the weight of target selection becomes even greater. The option of comparing a product with alternative therapies and maintaining reimbursement through price cuts will disappear.Another industry official said, “It is difficult to predict how things will unfold next year. Depending on the new implementation criteria, the number of targets could either increase or decrease. However, considering the practical work involved in reassessment, the authorities cannot simply increase the number of ingredients indiscriminately, so the annual number of reassessment targets may not change significantly.”
- Policy
- Prices of top-priced generics could drop 32% upon pricing reform
- by Chon, Seung-Hyun Mar 18, 2026 09:11am
- The government’s announced tiered pricing system for generics is expected to function as a powerful mechanism that significantly lowers generic drug prices. Concerns have been raised that the faster exposure to the tiered pricing system compared to the current system could dampen the momentum for later-entering generics. It is estimated that the price of the 13th generic to enter the market will drop to half the current level, as the discount rate increases for products that fail to meet the highest-price criteria, such as those failing bioequivalence tests.Analysis suggests that even if a product meets the highest-price criteria, if more than 13 products are launched simultaneously, a new price reduction mechanism, which applies an additional 15% price cut one year later, will be triggered, causing the price of the first generic to fall by 32% compared to current levels.Tiered pricing system with 15% reduction applied from the 13th generic onward... Drug prices plummet due to stricter eligibility requirementsAccording to industry sources on the 16th, the Ministry of Health and Welfare presented a principle at the HIPDC subcommittee meeting held on the 11th that, under the price reform, the 13th generic would be subject to pricing. The price reduction rate applied to each tier would be 15%.The price drop for generics become greater with the reformThe tiered pricing system is structured such that the insurance ceiling price decreases on a monthly basis, the later a generic enters the market. Although it was abolished in 2012, the system was reinstated with the 2020 drug pricing reform. Under the current system, if there are more than 20 pre-listed products of the same formulation, the price of generics entering the market as latecomers is reduced by 15% at each step.When reporting the drug pricing system reform to the Health Insurance Policy Deliberation Committee on November 11 of last year, the Ministry of Health and Welfare proposed a policy to grant the first generic a price reduced by 5 percentage points (p) from the calculated price starting from the 11th listing of the same formulation. This effectively means the Ministry presented a relaxed version of the stepped pricing system just four months after its initial report.However, because the tiered system would now begin applying from the 13th generic instead of the 21st under the current system, products would be exposed much earlier to this additional price-cutting mechanism.The impact of the tiered pricing system becomes even greater as the generic pricing benchmark itself is lowered. The government has proposed lowering the benchmark for calculating generic drug prices from the current 53.55% to the low to mid-40% range, a reduction of about 10 percentage points. If the generic drug pricing benchmark drops from 53.55% to 43%, the maximum generic price would be reduced by 19.7% in absolute terms.For example, under the current drug pricing system, when the maximum generic price is KRW 53.55, the 21st generic’s price cannot exceed KRW 45.52, which is a 15% reduction. The price of 22nd and 23rd generics then drops to KRW 38.69 and KRW 32.89, respectively. The 24th generic would be KRW 27.95, and the 25th would be KRW 23.76, meaning drug prices decrease as generics enter the market later.Under the revised pricing system, where the generic drug pricing benchmark is set at 43%, if the maximum price is KRW 43, the price of the 13th and 14th generics drops to KRW 36.55 and KRW 31.07, respectively, following a 15% step-down reduction. The price of the 15th generic drops to KRW 26.4. Under the current drug pricing system, the 15th generic drug is not subject to the tiered pricing structure and can maintain a price of 53.55%, but under the revised system, its price drops to less than half that level.As the stricter maximum price requirements under the revised system will also be applied, the magnitude of price reductions under the tiered pricing system will become even greater.Since July 2020, under the revised generic pricing rules, a generic product must both conduct its own bioequivalence study and use a registered active pharmaceutical ingredient to qualify for the top price. For each unmet requirement, the ceiling price is reduced by 15%. If both requirements are not met, the price falls by 27.75%. Applying that 15% reduction, the 53.55% premium benchmark falls to 45.52% if one requirement is unmet and to 38.69% if both are unmet.According to the criteria introduced by the ministry after the 2020 pricing reform, “Even if a product meets both premium-price requirements, if there are already 20 or more listed identical products, starting from the 21st product, the price will be listed at 85% of the lower of the lowest price of the identical product or 38.69%.” The 38.69% rate is the result of two 15% reductions applied when a product failed to meet both of the highest-price criteria. (53.55% × 0.85 × 0.85)When 20 or more generics are listed, the 21st drug is listed at the lowest price vs 38.69%, whichever is lowerCurrently, the 21st generic drug,the first to be subject to the tiered drug pricing system, is priced at 32.86%, which is a 15% reduction from the 38.69% baseline. Compared to the highest price of 53.33%, this means the first generic drug subject to the tiered pricing system will see a 38.6% reduction. The prices of the 22nd and 23rd generic drugs will be reduced even further.The Ministry of Health and Welfare plans to increase the reduction rate applied when the highest price requirement is not met under the revised drug pricing system from 15% to 20%. If the benchmark for the highest generic drug price is set at 43%, the calculation standard will be further lowered to 34.40% for generics that fail to meet one requirement, and to 27.52% for generics that fail to meet both requirements.Under the revised drug pricing system’s tiered pricing structure, which applies the highest-price requirement, the ceiling price drops significantly starting with the 13th generic.The price of the 13th generic is calculated to drop from 27.52%, the rate applied to generics failing to meet two highest-price requirements, to 23.39%, a reduction of 15%. Comparing the same 13th generic drug, the price under the current system is KRW 53.55, whereas under the revised system, it drops to roughly half that amount. The 13th and 14th generic drugs each decrease by 38.6% from the lowest price, falling to KRW 14.36 and KRW 8.8, respectively.Under the revised drug pricing reform, the combined effects of shortening the sequence for applying the tiered pricing system, a 15% reduction upon tiered application, and a 20% price reduction for drugs that do not meet the highest-price requirement create a structure that effectively prevents late-entrant generics from entering the market.If 13 or more highest-priced generics are listed, prices will be reduced by 15% after one year.Even top-priced generics could see their prices cut a year later, depending on the number of products listed at the same time.To prevent excessive competition when the first generics enter, the ministry plans to apply pricing standards equivalent to tiered cuts to generics whose listing causes the total number of identical products to exceed 13.Even if a generic is listed within the first 12 products and initially receives the top price of 43%, if that product is among a group whose simultaneous entry causes the total to exceed 13, its price would be cut by 15% one year later.The price of generics with many listings will fall to a greater extent with the new reformFor example, if 8 generic products are listed in January and receive prices in the low-to-mid 40% range, and another 8 generics are listed in February, those products would all be treated as being listed simultaneously starting from the 9th position and could receive the top price. However, because the additional eight February listings push the total number of identical products beyond 13, those products would, one year later, fall to the 85% tiered level.Even the earliest listed generic could have its price cut by 15% one year later if 13 or more products enter simultaneously. In other words, even if it initially secures the top-price benchmark of 43%, it could later fall to 36.55% after one year. In that case, the price would be 31.75% lower than the current ceiling price.Within the industry, this is being interpreted as the government effectively intending to allow only up to 12 generics. Under restrictions on joint development, only three products may participate in one bioequivalence study. Generics beyond three such groups would face such sharp price declines that their incentive to enter the market would effectively collapse.One industry official noted, “Once the revised drug pricing system is implemented, late-entering generics will effectively be unable to turn a profit, so competition to secure the highest price by capturing the market first will inevitably intensify. Even if a company secures a leading position in the generic market, if there are many competing products, the tiered pricing mechanism will cause drug prices to fall, which could even lead to companies abandoning their plans to enter the generic market altogether.”