




-
 JW중외제약, 통풍 신약 국내 3상 임상계획 승인[데일리팜=정새임 기자] JW중외제약은 24일 식품의약처안전처로부터 통풍 치료제 후보 물질 'URC-102' 3상 임상시험계획을 승인받았다고 공시했다. URC-102 3상은 전체 588명 통풍 환자를 대상으로 하는 다국가·다기관 무작위배정, 이중눈가림 임상이다. 국내에선 165명 환자를 대상으로 한다. 한국 외 대만과 유럽에도 임상시험계획을 제출할 예정이다. 3상은 URC-102 투여 후 혈중요산 감소 효과가 기존 치료제인 페북소스타트 대비 비열등함을 입증하는 것이 목표다. 3상 임상 종료 예정일은 2025년 12월이다. URC-102는 요산이 우리 몸에 다시 흡수되도록 하는 요산 트랜스포터(URAT)-1을 억제하는 기전의 요산 배설 촉진제다. 혈액 내 요산 농도가 비정상적으로 높은 고요산혈증으로 인한 통풍질환에 유효하다. JW중외제약은 "기존 치료제인 알로푸리놀은 전 세계적으로 많이 쓰이지만 효과가 약한 편이고, 특히 동양인에서 알레르기 우려가 있다. 다른 약제인 페북소스타트는 안전성 우려로 미국에서 1차 치료제로 쓰이지 못하고 있다"며 URC-102는 2상에서 안전성 우려가 낮고 효과가 확인됨에 따라 3상 확증임상에서 유효성과 안전성을 입증하겠다"고 밝혔다.2022-11-24 18:18:10정새임
JW중외제약, 통풍 신약 국내 3상 임상계획 승인[데일리팜=정새임 기자] JW중외제약은 24일 식품의약처안전처로부터 통풍 치료제 후보 물질 'URC-102' 3상 임상시험계획을 승인받았다고 공시했다. URC-102 3상은 전체 588명 통풍 환자를 대상으로 하는 다국가·다기관 무작위배정, 이중눈가림 임상이다. 국내에선 165명 환자를 대상으로 한다. 한국 외 대만과 유럽에도 임상시험계획을 제출할 예정이다. 3상은 URC-102 투여 후 혈중요산 감소 효과가 기존 치료제인 페북소스타트 대비 비열등함을 입증하는 것이 목표다. 3상 임상 종료 예정일은 2025년 12월이다. URC-102는 요산이 우리 몸에 다시 흡수되도록 하는 요산 트랜스포터(URAT)-1을 억제하는 기전의 요산 배설 촉진제다. 혈액 내 요산 농도가 비정상적으로 높은 고요산혈증으로 인한 통풍질환에 유효하다. JW중외제약은 "기존 치료제인 알로푸리놀은 전 세계적으로 많이 쓰이지만 효과가 약한 편이고, 특히 동양인에서 알레르기 우려가 있다. 다른 약제인 페북소스타트는 안전성 우려로 미국에서 1차 치료제로 쓰이지 못하고 있다"며 URC-102는 2상에서 안전성 우려가 낮고 효과가 확인됨에 따라 3상 확증임상에서 유효성과 안전성을 입증하겠다"고 밝혔다.2022-11-24 18:18:10정새임 -
파마킹, 비알코올성 지방간 치료제 3상 환자 등록 완료[데일리팜=노병철 기자] 파마킹은 비알코올성 지방간(NAFLD) 치료 후보물질 PMK-N01GI1의 임상3상 환자 등록이 지난 달 완료됐다고 24일 밝혔다. 회사 측은 모집 환자수가 144명을 넘어 통계적 유의성 확보가 가능하다는 판단이다. PMK-N01GI1 3상은 간경변을 제외한 NAFLD 환자들을 대상으로 국내 21개 대학병원에서 진행되고 있다. NAFLD는 유의한 알코올 섭취, 지방간을 초래하는 약물의 복용, 동반된 다른 원인에 의한 간질환 등이 없으면서 영상의학 검사나 조직검사에서 간 내 지방침착의 소견을 보이는 질환으로 비알코올성 지방간, 비알코올성 지방간염으로 연관된 간경변증을 포괄하는 진단명이다. 하지만 현재 지방간을 약물학적으로 치료하는데 유용한 약제는 거의 없는 상태이며 운동과 식이용법만이 권장되고 있다. 실제로 이러한 방법에 의한 지방간의 치료 효율은 매우 낮아서 전 세계적으로 유효한 치료제 개발에 대한 요구가 절실한 상황이다. 파마킹은 후보물질 PMK-N01GI1이 간에 축적된 중성지방을 낮추는 치료제로서의 가능성이 있다고 판단, 2011년부터 개발에 들어갔다. 회사 측이 밝힌 최종 결과 도출 시점은 2023년 상반기 중이다.2022-11-24 08:23:01노병철
-
 코로나 신약 개발해도...실제 성과 내기엔 '산 넘어 산'[데일리팜=김진구 기자] SK바이오사이언스가 자체 개발한 코로나19 백신 스카이코비원의 완제 생산라인이 멈춰 섰다. 회사는 원액 생산을 지속하면서 정부의 추가 요청을 기다린다는 입장이지만, 현재로선 생산 재개 가능성이 떨어진다는 전망이 지배적이다. 지난해와 달리 백신 접종률이 크게 하락했기 때문이다. 어렵게 코로나 백신 개발에 성공했으나, 팬데믹 사태의 끝자락에 제품이 공급되면서 기대했던 만큼의 성과를 내지 못하고 있다는 평가다. 제약업계의 관심은 일본에서 긴급사용승인을 받은 경구용 코로나 치료제 조코바로 쏠린다. 일동제약과 시오노기제약이 공동 개발 중인 이 치료제가 국내 허가기관의 문턱을 넘을 경우 얼마나 성공을 거둘지에 대해 물음표가 붙는 상황이다. ◆접종률 97%→5% 뚝…SK바사 '생산 잠정 중단' 결정 SK바이오사이언스는 지난 23일 코로나19 백신 스카이코비원의 완제 생산이 중단된 상태라고 공시했다. SK바이오사이언스는 지난 6월 국산 1호 코로나 백신으로 스카이코비원을 허가 받았다. 9월엔 정부와 선구매 계약에 따라 1000만 도즈 중 60만 도즈를 초도 물량으로 공급했다. 다만 이후로는 추가 생산·공급이 중단된 상태다. 코로나 예방접종률이 낮아지면서 백신 수요가 급감했기 때문이다. 질병관리청은 지난달 10일 코로나 백신의 동절기 추가 접종을 권고했다. 그러나 23일 기준 동절기 추가 접종률은 4.5%에 그친다. 누적 230만9305명이 백신을 접종했다. 지난해의 경우 4400만명 이상이 백신을 접종했다. 현재 전국민 기초 접종률은 87.1%에 달한다. 경쟁 백신에 비해 시장 진입이 늦었다는 점도 스카이코비원의 생산 중단 이유로 설명된다. 실제 백신별 동절기 추가 접종은 모더나 백신 153만332명, 화이자 백신 75만4058명, 노바백스 백신 2만3156명 등이다. 스카이코비원은 1759명에 그친다. 초도 물량으로 공급한 60만 도즈 대부분이 잔여물량으로 남아있는 셈이다. 올해 초 공급을 중단한 국산 코로나 치료제 렉키로나와는 대조적이다. 셀트리온은 지난해 2월 항체치료제로 렉키로나를 조건부 허가 받았다. 9월엔 정식 허가를 받았다. 당시엔 코로나 치료제로 길리어드의 렘데시비르가 유일했다. 코로나 백신은 개발되기 전이었다. 코로나 바이러스의 감염 전파력과 치명률도 지금보다 높았다. 렉키로나는 지난해 코로나 환자 5만명에게 투여됐다. 지난해 생산실적은 1895억원에 달한다. 다만 셀트리온은 올해 2월 렉키로나의 신규 공급 중단을 결정했다. 오미크론 변이 바이러스 확대와 경구용 코로나 치료제 등장이 복합적으로 영향을 끼쳤다. 비록 공급이 중단됐지만 렉키로나는 비교적 빠르게 시장에 진입하면서 적잖은 성공을 거뒀다는 평가다. ◆긴급사용승인 문턱 '조코바' 시장 성공 거둘까 제약업계의 관심은 일동제약과 일본 시오노기제약이 공동 개발 중인 경구용 코로나19 치료제 조코바로 향한다. 여러모로 스카이코비원과 사정이 비슷하기 때문이다. 일본 후생노동성은 지난 22일 조코바의 긴급사용승인을 권고했다. 조코바가 일본 규제기관의 문턱을 넘으면서 한국에서의 긴급사용승인 가능성도 높아지고 있다. 일동제약은 지난 8월 국내 임상2/3상을 마무리하고 임상 종료 보고서를 식품의약품안전처에 제출한 상태다. 다만 조코바가 국내에서 긴급사용승인을 받더라도 상업적 성공까지 이어질지에 대해선 의문부호가 붙는다. 스카이코비원 사례와 마찬가지로 시장 진입이 경쟁 제품에 비해 늦기 때문이다. 국내에서 경구용 코로나19 치료제로는 화이자 팍스로비드가 작년 12월, MSD 라게브리오가 올해 3월 각각 긴급사용승인을 받았다. 경구용 코로나19 치료제가 당초 기대와 달리 처방률이 높지 않다는 점도 조코바의 성공을 낙관하기 어려운 이유로 꼽힌다. 팍스로비드와 라게브리오는 긴급사용승인 당시만 하더라도 '게임체인저'로 주목을 받았다. 그러나 현재는 낮은 처방률로 고민하고 있다. 질병청에 따르면 10월 마지막 주 기준 60세 이상 고령층의 경구용 코로나19 치료제 처방률은 31.7%에 그친다. 경구용 코로나19 치료제 처방률은 9월 이후 20%대 후반과 30%대 초반 사이에서 정체돼 있다. 경구용 코로나19 치료제는 사실상 감기약에 밀렸다는 평가다. 코로나 바이러스는 몇 차례 변이를 거치면서 치명률이 낮아졌다. 대부분 경증 환자는 감기약만으로 코로나19 증상이 완화된다. 이런 이유로 지난 3월 코로나 재유행 당시 정부는 경증환자에게 감기약 처방을 권고했다. 이후로 감기약 수요가 급증했고 수급난은 현재까지 이어지고 있다. 다른 코로나19 백신·치료제 개발 업체들도 비슷한 고민을 안고 있다. 현재 코로나19 치료제는 신풍제약, 현대바이오사이언스, 텔콘RF제약, 아미코젠파마, 녹십자웰빙, 대원제약, 진원생명과학, 유나이티드, 이뮨메드, 동화약품, 크리스탈지노믹스, 제넨셀 등이 개발 중이다. GC녹십자와 일양약품, 부광약품은 코로나 치료제 개발을 포기했다. 코로나19 백신은 셀리드, 유바이오로직스, 진원생명과학, 큐라티스, 아이진 등이 개발하고 있다. 제넥신과 HK이노엔은 개발을 중단했다.2022-11-24 06:20:26김진구
코로나 신약 개발해도...실제 성과 내기엔 '산 넘어 산'[데일리팜=김진구 기자] SK바이오사이언스가 자체 개발한 코로나19 백신 스카이코비원의 완제 생산라인이 멈춰 섰다. 회사는 원액 생산을 지속하면서 정부의 추가 요청을 기다린다는 입장이지만, 현재로선 생산 재개 가능성이 떨어진다는 전망이 지배적이다. 지난해와 달리 백신 접종률이 크게 하락했기 때문이다. 어렵게 코로나 백신 개발에 성공했으나, 팬데믹 사태의 끝자락에 제품이 공급되면서 기대했던 만큼의 성과를 내지 못하고 있다는 평가다. 제약업계의 관심은 일본에서 긴급사용승인을 받은 경구용 코로나 치료제 조코바로 쏠린다. 일동제약과 시오노기제약이 공동 개발 중인 이 치료제가 국내 허가기관의 문턱을 넘을 경우 얼마나 성공을 거둘지에 대해 물음표가 붙는 상황이다. ◆접종률 97%→5% 뚝…SK바사 '생산 잠정 중단' 결정 SK바이오사이언스는 지난 23일 코로나19 백신 스카이코비원의 완제 생산이 중단된 상태라고 공시했다. SK바이오사이언스는 지난 6월 국산 1호 코로나 백신으로 스카이코비원을 허가 받았다. 9월엔 정부와 선구매 계약에 따라 1000만 도즈 중 60만 도즈를 초도 물량으로 공급했다. 다만 이후로는 추가 생산·공급이 중단된 상태다. 코로나 예방접종률이 낮아지면서 백신 수요가 급감했기 때문이다. 질병관리청은 지난달 10일 코로나 백신의 동절기 추가 접종을 권고했다. 그러나 23일 기준 동절기 추가 접종률은 4.5%에 그친다. 누적 230만9305명이 백신을 접종했다. 지난해의 경우 4400만명 이상이 백신을 접종했다. 현재 전국민 기초 접종률은 87.1%에 달한다. 경쟁 백신에 비해 시장 진입이 늦었다는 점도 스카이코비원의 생산 중단 이유로 설명된다. 실제 백신별 동절기 추가 접종은 모더나 백신 153만332명, 화이자 백신 75만4058명, 노바백스 백신 2만3156명 등이다. 스카이코비원은 1759명에 그친다. 초도 물량으로 공급한 60만 도즈 대부분이 잔여물량으로 남아있는 셈이다. 올해 초 공급을 중단한 국산 코로나 치료제 렉키로나와는 대조적이다. 셀트리온은 지난해 2월 항체치료제로 렉키로나를 조건부 허가 받았다. 9월엔 정식 허가를 받았다. 당시엔 코로나 치료제로 길리어드의 렘데시비르가 유일했다. 코로나 백신은 개발되기 전이었다. 코로나 바이러스의 감염 전파력과 치명률도 지금보다 높았다. 렉키로나는 지난해 코로나 환자 5만명에게 투여됐다. 지난해 생산실적은 1895억원에 달한다. 다만 셀트리온은 올해 2월 렉키로나의 신규 공급 중단을 결정했다. 오미크론 변이 바이러스 확대와 경구용 코로나 치료제 등장이 복합적으로 영향을 끼쳤다. 비록 공급이 중단됐지만 렉키로나는 비교적 빠르게 시장에 진입하면서 적잖은 성공을 거뒀다는 평가다. ◆긴급사용승인 문턱 '조코바' 시장 성공 거둘까 제약업계의 관심은 일동제약과 일본 시오노기제약이 공동 개발 중인 경구용 코로나19 치료제 조코바로 향한다. 여러모로 스카이코비원과 사정이 비슷하기 때문이다. 일본 후생노동성은 지난 22일 조코바의 긴급사용승인을 권고했다. 조코바가 일본 규제기관의 문턱을 넘으면서 한국에서의 긴급사용승인 가능성도 높아지고 있다. 일동제약은 지난 8월 국내 임상2/3상을 마무리하고 임상 종료 보고서를 식품의약품안전처에 제출한 상태다. 다만 조코바가 국내에서 긴급사용승인을 받더라도 상업적 성공까지 이어질지에 대해선 의문부호가 붙는다. 스카이코비원 사례와 마찬가지로 시장 진입이 경쟁 제품에 비해 늦기 때문이다. 국내에서 경구용 코로나19 치료제로는 화이자 팍스로비드가 작년 12월, MSD 라게브리오가 올해 3월 각각 긴급사용승인을 받았다. 경구용 코로나19 치료제가 당초 기대와 달리 처방률이 높지 않다는 점도 조코바의 성공을 낙관하기 어려운 이유로 꼽힌다. 팍스로비드와 라게브리오는 긴급사용승인 당시만 하더라도 '게임체인저'로 주목을 받았다. 그러나 현재는 낮은 처방률로 고민하고 있다. 질병청에 따르면 10월 마지막 주 기준 60세 이상 고령층의 경구용 코로나19 치료제 처방률은 31.7%에 그친다. 경구용 코로나19 치료제 처방률은 9월 이후 20%대 후반과 30%대 초반 사이에서 정체돼 있다. 경구용 코로나19 치료제는 사실상 감기약에 밀렸다는 평가다. 코로나 바이러스는 몇 차례 변이를 거치면서 치명률이 낮아졌다. 대부분 경증 환자는 감기약만으로 코로나19 증상이 완화된다. 이런 이유로 지난 3월 코로나 재유행 당시 정부는 경증환자에게 감기약 처방을 권고했다. 이후로 감기약 수요가 급증했고 수급난은 현재까지 이어지고 있다. 다른 코로나19 백신·치료제 개발 업체들도 비슷한 고민을 안고 있다. 현재 코로나19 치료제는 신풍제약, 현대바이오사이언스, 텔콘RF제약, 아미코젠파마, 녹십자웰빙, 대원제약, 진원생명과학, 유나이티드, 이뮨메드, 동화약품, 크리스탈지노믹스, 제넨셀 등이 개발 중이다. GC녹십자와 일양약품, 부광약품은 코로나 치료제 개발을 포기했다. 코로나19 백신은 셀리드, 유바이오로직스, 진원생명과학, 큐라티스, 아이진 등이 개발하고 있다. 제넥신과 HK이노엔은 개발을 중단했다.2022-11-24 06:20:26김진구 -
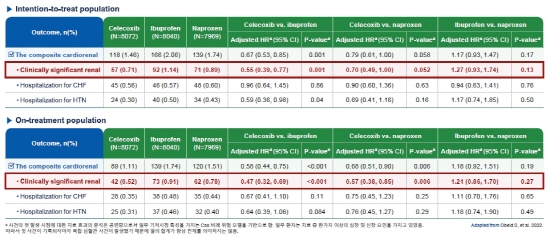 진통제 '쎄레브렉스', 신장과 심혈관계 안전성 재확인[데일리팜=어윤호 기자] COX-2억제제 '쎄레브렉스'가 신장과 심혈관계 안전성을 재차 입증했다. 비아트리스는 'PRECISION(Prospective Randomized Evaluation of Celecoxib Integrated Safety vs. Ibuprofen Or Naproxen)-Renal' 연구를 통해 쎄레브렉스(세레콕시브)와 이부프로펜, 나프록센 등 진통제들과 비교해 다양한 안전성 프로파일을 확보했다. 해당 연구 결과는 지난 3월 유럽심장학회(ESC, European Society Cardiology, ESC) 학술지 '유럽심장저널-심혈관 약물요법(European Heart Journal - Cardiovascular Pharmacotherapy)'에 게재됐으며 최근 최종 수정을 마쳤다. PRECISION 연구는 심혈관계 고위험의 골관절염 또는 류마티스관절염 환자를 대상으로 쎄레브렉스의 신장, 심혈관계, 위장관계 안전성을 살펴본 연구이다. 올해 ESC에서 발표된 PRECISION-Renal 연구에서 치료 의향(intention-to-treat) 환자 분석 결과, 쎄레브렉스는 임상적으로 유의한 신장 사건의 발생률이 이부프로펜 대비 유의하게 낮았으며 나프록센과는 유의한 차이를 보이지 않았다. 1차 심장 및 신장 복합 결과(Primary Cardiorenal Composite Outcome)의 발생 위험은 쎄레브렉스가 이부프로펜 대비 33% 유의하게 낮았으며 나프록센 대비 21% 더 낮은 경향을 보였다. 치료 의향 환자 분석에 이어 치료 완료(on-treatment) 환자 분석 결과에서는 임상적으로 유의한 신장 사건의 발생률이 쎄레브렉스가 이부프로펜 및 나프록센 대비 유의하게 낮았다. 1차 심장 및 신장 복합 결과의 발생 위험 또한 쎄레브렉스가 이부프로펜 및 나프록센 대비 유의하게 낮은 것으로 나타났다. 한편 쎄레브렉스는 2000년 65세 이상 골관절염 및 류마티스관절염 환자를 대상으로 국내 식품의약품안전처 허가를 받았다. 이후 2017년 모든 성인의 골관절염, 류마티스관절염, 강직성 척추염 환자로 적응증이 확대되었으며, 현재 100mg, 200mg은 국내에서 골관절염, 류마티스관절염, 강직성 척추염의 증상 및 징후 완화와 성인의 급성 통증 개선, 원발월경통 치료에 사용되고 있다.2022-11-23 12:18:01어윤호
진통제 '쎄레브렉스', 신장과 심혈관계 안전성 재확인[데일리팜=어윤호 기자] COX-2억제제 '쎄레브렉스'가 신장과 심혈관계 안전성을 재차 입증했다. 비아트리스는 'PRECISION(Prospective Randomized Evaluation of Celecoxib Integrated Safety vs. Ibuprofen Or Naproxen)-Renal' 연구를 통해 쎄레브렉스(세레콕시브)와 이부프로펜, 나프록센 등 진통제들과 비교해 다양한 안전성 프로파일을 확보했다. 해당 연구 결과는 지난 3월 유럽심장학회(ESC, European Society Cardiology, ESC) 학술지 '유럽심장저널-심혈관 약물요법(European Heart Journal - Cardiovascular Pharmacotherapy)'에 게재됐으며 최근 최종 수정을 마쳤다. PRECISION 연구는 심혈관계 고위험의 골관절염 또는 류마티스관절염 환자를 대상으로 쎄레브렉스의 신장, 심혈관계, 위장관계 안전성을 살펴본 연구이다. 올해 ESC에서 발표된 PRECISION-Renal 연구에서 치료 의향(intention-to-treat) 환자 분석 결과, 쎄레브렉스는 임상적으로 유의한 신장 사건의 발생률이 이부프로펜 대비 유의하게 낮았으며 나프록센과는 유의한 차이를 보이지 않았다. 1차 심장 및 신장 복합 결과(Primary Cardiorenal Composite Outcome)의 발생 위험은 쎄레브렉스가 이부프로펜 대비 33% 유의하게 낮았으며 나프록센 대비 21% 더 낮은 경향을 보였다. 치료 의향 환자 분석에 이어 치료 완료(on-treatment) 환자 분석 결과에서는 임상적으로 유의한 신장 사건의 발생률이 쎄레브렉스가 이부프로펜 및 나프록센 대비 유의하게 낮았다. 1차 심장 및 신장 복합 결과의 발생 위험 또한 쎄레브렉스가 이부프로펜 및 나프록센 대비 유의하게 낮은 것으로 나타났다. 한편 쎄레브렉스는 2000년 65세 이상 골관절염 및 류마티스관절염 환자를 대상으로 국내 식품의약품안전처 허가를 받았다. 이후 2017년 모든 성인의 골관절염, 류마티스관절염, 강직성 척추염 환자로 적응증이 확대되었으며, 현재 100mg, 200mg은 국내에서 골관절염, 류마티스관절염, 강직성 척추염의 증상 및 징후 완화와 성인의 급성 통증 개선, 원발월경통 치료에 사용되고 있다.2022-11-23 12:18:01어윤호 -
 종근당 "루센비에스, 오리지널과 동등"...국제학술지 게재[데일리팜=천승현 기자] 종근당은 황반변성치료제 ‘루센티스’ 바이오시밀러 ‘루센비에스’의 임상 3상 결과가 SCI급 국제학술지 ‘플로스원(PLoS One)’에 게재됐다고 23일 밝혔다. 종근당은 2018년 9월부터 2021년 3월까지 서울대학교병원을 비롯한 25개 병원에서 총 312명의 신생혈관성(습성) 연령관련 황반변성 환자를 대상으로 루센비에스의 임상 3상을 진행했다. 임상 3상에서 약물투여 후 3개월 시점의 최대교정시력(BCVA)을 비교하는 1차 유효성 평가지표에서 15글자 미만의 시력 손실을 보인 환자의 비율을 분석한 결과 루센비에스 투여군 97.95%(143/146명), 오리지널 약물 투여군 98.62%(143/145명)으로 두 약물간 동등성 범위를 충족했다. 최대교정시력(BCVA)의 변화는 루센비에스 투여군 7.14글자, 오리지널 투여군 6.28글자가 개선돼 두 약물 간 유의한 차이를 보이지 않았다. 약물 투여 후 3, 6, 12개월이 경과한 시점에서 각각 15글자 미만의 시력 손실 및 시력 호전을 보인 환자의 비율과 중심망막두께 변화 등의 지표를 통해 약물 효능 및 안전성, 면역원성, 약동학적 특성 모두 오리지널 약물과 임상적으로 동등함을 확인했다. 지난달 식품의약품안전처 허가를 받은 루센비에스는 라니비주맙을 주성분으로 하는 고순도의 루센티스 바이오시밀러다. 종근당의 순수 독자 기술인 항체절편 원료제조 기술이 적용됐다. 임상 논문 책임저자인 유형곤 교수는 “루센비에스는 1차 평가지표인 최대교정시력을 비롯해 주요 지표에서 오리지널 약물인 루센티스 대비 임상적 동등성이 검증된 약물”이라며 “환자의 상태에 맞춘 선택적 투여 요법(PRN, Pro Re Nata)으로 황반변성 질환의 상태를 안정화할 뿐만 아니라 고정적 약물 투여로 인한 환자의 치료 부담을 줄일 수 있을 것으로 기대한다” 고 말했다. 플로스원은 플로스(PLoS, Public Library Of Science)에서 2006년부터 발행하는 과학 및 의약 분야 전문 온라인 학술지다. 지난 5년 연속 글로벌 학술논문 데이터베이스 플랫폼인 SCIE와 스코퍼스(SCOPUS)에 등재된 바 있다.2022-11-23 10:06:49천승현
종근당 "루센비에스, 오리지널과 동등"...국제학술지 게재[데일리팜=천승현 기자] 종근당은 황반변성치료제 ‘루센티스’ 바이오시밀러 ‘루센비에스’의 임상 3상 결과가 SCI급 국제학술지 ‘플로스원(PLoS One)’에 게재됐다고 23일 밝혔다. 종근당은 2018년 9월부터 2021년 3월까지 서울대학교병원을 비롯한 25개 병원에서 총 312명의 신생혈관성(습성) 연령관련 황반변성 환자를 대상으로 루센비에스의 임상 3상을 진행했다. 임상 3상에서 약물투여 후 3개월 시점의 최대교정시력(BCVA)을 비교하는 1차 유효성 평가지표에서 15글자 미만의 시력 손실을 보인 환자의 비율을 분석한 결과 루센비에스 투여군 97.95%(143/146명), 오리지널 약물 투여군 98.62%(143/145명)으로 두 약물간 동등성 범위를 충족했다. 최대교정시력(BCVA)의 변화는 루센비에스 투여군 7.14글자, 오리지널 투여군 6.28글자가 개선돼 두 약물 간 유의한 차이를 보이지 않았다. 약물 투여 후 3, 6, 12개월이 경과한 시점에서 각각 15글자 미만의 시력 손실 및 시력 호전을 보인 환자의 비율과 중심망막두께 변화 등의 지표를 통해 약물 효능 및 안전성, 면역원성, 약동학적 특성 모두 오리지널 약물과 임상적으로 동등함을 확인했다. 지난달 식품의약품안전처 허가를 받은 루센비에스는 라니비주맙을 주성분으로 하는 고순도의 루센티스 바이오시밀러다. 종근당의 순수 독자 기술인 항체절편 원료제조 기술이 적용됐다. 임상 논문 책임저자인 유형곤 교수는 “루센비에스는 1차 평가지표인 최대교정시력을 비롯해 주요 지표에서 오리지널 약물인 루센티스 대비 임상적 동등성이 검증된 약물”이라며 “환자의 상태에 맞춘 선택적 투여 요법(PRN, Pro Re Nata)으로 황반변성 질환의 상태를 안정화할 뿐만 아니라 고정적 약물 투여로 인한 환자의 치료 부담을 줄일 수 있을 것으로 기대한다” 고 말했다. 플로스원은 플로스(PLoS, Public Library Of Science)에서 2006년부터 발행하는 과학 및 의약 분야 전문 온라인 학술지다. 지난 5년 연속 글로벌 학술논문 데이터베이스 플랫폼인 SCIE와 스코퍼스(SCOPUS)에 등재된 바 있다.2022-11-23 10:06:49천승현 -
 일동제약 "시오노기, 코로나치료제 日 긴급사용 승인"[데일리팜=천승현 기자] 일동제약은 일본 후생노동성이 시오노기제약의 경구용 코로나19 치료제 조코바(S-S-217622)의 긴급사용을 승인했다고 23일 공시했다. 후생노동성 약사·식품위생심의회는 조코바가 긴급승인제도 적용에 부합한다고 논의했고 일본 후생노동성이 긴급사용승인을 발표했다. 앞서 시오노기제약은 지난 2월 25일 일본 후생노동성에 조코바의 조건부허가를 신청했다. 하지만 지난 6월과 7월 두 차례 ‘데이터가 충분치 않다'는 이유로 보류 판정을 받았다. 시오노기제약이 지난 9월 홈페이지를 통해 공개한 조코바 임상 3상 결과 한국·일본·베트남의 경증·중등증 환자 1821명을 대상으로 진행된 임상에서 조코바를 투여한 환자는 코로나 주요 5개 증상(코막힘·콧물·인후통·기침·발열)이 가라앉을 때까지 시간이 위약을 투여한 환자에 비해 유의하게 낮은 것으로 나타났다. 증상 억제까지 시간은 저용량 엔시트렐비르를 투여한 그룹이 167.9시간(약 7일)이었고, 위약을 투여한 그룹은 192.2시간(약 8일)이었다. 투여 4일차에 바이러스 RNA가 얼마나 경감됐는지 살핀 결과에서도 엔시트렐비르 투여군은 위약 대비 유의하게 감소한 것으로 나타났다. 조코바는 일동제약이 국내 판권을 보유한 제품이다. 일동제약은 지난해 11월 일본 시오노기제약의 경구용 코로나 치료제 후보물질 ‘S-217622’에 대한 국내 임상에 돌입했다. 일동제약은 지난 8월 국내 임상2/3상을 마무리하고, 임상 종료 보고서를 식품의약품안전처에 제출했다. 지난 9월 시오노기제약과 라이선스인 계약을 체결하며 국내 판권을 확보했다.2022-11-23 07:51:32천승현
일동제약 "시오노기, 코로나치료제 日 긴급사용 승인"[데일리팜=천승현 기자] 일동제약은 일본 후생노동성이 시오노기제약의 경구용 코로나19 치료제 조코바(S-S-217622)의 긴급사용을 승인했다고 23일 공시했다. 후생노동성 약사·식품위생심의회는 조코바가 긴급승인제도 적용에 부합한다고 논의했고 일본 후생노동성이 긴급사용승인을 발표했다. 앞서 시오노기제약은 지난 2월 25일 일본 후생노동성에 조코바의 조건부허가를 신청했다. 하지만 지난 6월과 7월 두 차례 ‘데이터가 충분치 않다'는 이유로 보류 판정을 받았다. 시오노기제약이 지난 9월 홈페이지를 통해 공개한 조코바 임상 3상 결과 한국·일본·베트남의 경증·중등증 환자 1821명을 대상으로 진행된 임상에서 조코바를 투여한 환자는 코로나 주요 5개 증상(코막힘·콧물·인후통·기침·발열)이 가라앉을 때까지 시간이 위약을 투여한 환자에 비해 유의하게 낮은 것으로 나타났다. 증상 억제까지 시간은 저용량 엔시트렐비르를 투여한 그룹이 167.9시간(약 7일)이었고, 위약을 투여한 그룹은 192.2시간(약 8일)이었다. 투여 4일차에 바이러스 RNA가 얼마나 경감됐는지 살핀 결과에서도 엔시트렐비르 투여군은 위약 대비 유의하게 감소한 것으로 나타났다. 조코바는 일동제약이 국내 판권을 보유한 제품이다. 일동제약은 지난해 11월 일본 시오노기제약의 경구용 코로나 치료제 후보물질 ‘S-217622’에 대한 국내 임상에 돌입했다. 일동제약은 지난 8월 국내 임상2/3상을 마무리하고, 임상 종료 보고서를 식품의약품안전처에 제출했다. 지난 9월 시오노기제약과 라이선스인 계약을 체결하며 국내 판권을 확보했다.2022-11-23 07:51:32천승현 -
 대웅제약 "SGLT-2 당뇨신약, 반려동물 장기 안전성 확인"[데일리팜=천승현 기자] 대웅제약은 당뇨병치료제로 개발 중인 ‘DWP16001’의 안전성이 반려동물 대상 연구에서 확인됐다고 22일 밝혔다. DWP16001은 이나보글리플로진 성분의 SGLT-2 억제제 계열 당뇨치료제다. 대웅제약은 지난 17일 대한수의학회에서 반려동물을 대상으로 DWP16001의 당뇨병 치료 효과에 대한 연구자 추가 임상 결과를 공개했다. 이번에 발표한 내용은 지난해 5월 발표한 8주 간의 연구자 임상에 참여했던 인슐린 의존성 당뇨병 반려견을 대상으로 1년 연장 투약에 대한 장기 안전성을 추가로 검증한 연구 결과다. 연구에서 인슐린과 DWP16001을 1년 동안 1일 1회 또는 3일 1회 병용투여하고, 각 군에게 혈중 케톤 및 젖산탈수소효소(Lactate dehydrogenase, LDH) 검사, 일반 혈액(Complete Blood Cell, CBC) 검사, 혈청화학(serum chemistry) 검사, 전해질 검사, 요검사 등을 시행했다. 혈중 케톤 및 LDH 검사는 ‘당뇨병성 케톤산증’ 평가 검사로 저혈당과 함께 가장 주요한 부작용 평가 지표로 알려졌다. 검사 결과 약물을 투여한 1년 동안 혈중 케톤 및 LDH의 유의적인 변화가 없었다. 당뇨 반려견 치료 및 관리에서 가장 중요한 부작용인 저혈당증 및 당뇨병성 케톤산증은 없는 것으로 확인됐다. 백혈구, 적혈구, 혈소판 및 주요 장기(간, 신장 등)에 대한 유의한 수치 변화는 확인되지 않았다. 대웅제약은 “당뇨 반려견 대상 인슐린 및 DWP16001 1년 장기 투약에 대한 장기 안전성을 확보하게 됐다”라고 평가했다. 이번 연구의 책임 연구자인 윤화영 서울대학교 수의과대학 교수는 “당뇨병 반려견에 인슐린 및 DWP16001 투약을 1년에 걸쳐 장기 적용해 본 결과, 안전성 및 혈당조절효과가 확인됐다”고 말했다. 대웅제약은 두 차례의 연구자 주도임상으로 유효성 및 안전성을 확인한 만큼 실제 동물의약품 출시로 이어질 수 있도록 개발을 가속화 한다는 계획이다. 전승호 대웅제약 대표는 "두 건의 연구자 임상으로 당뇨 반려견에서 DWP16001의 유효성 및 안전성을 모두 확인했다"며 "반려동물 대상 의약품으로 개발해 경구 치료제가 없는 반려동물 시장에서 새로운 선택지를 제시할 수 있을 것으로 기대한다"고 말했다.2022-11-22 09:45:41천승현
대웅제약 "SGLT-2 당뇨신약, 반려동물 장기 안전성 확인"[데일리팜=천승현 기자] 대웅제약은 당뇨병치료제로 개발 중인 ‘DWP16001’의 안전성이 반려동물 대상 연구에서 확인됐다고 22일 밝혔다. DWP16001은 이나보글리플로진 성분의 SGLT-2 억제제 계열 당뇨치료제다. 대웅제약은 지난 17일 대한수의학회에서 반려동물을 대상으로 DWP16001의 당뇨병 치료 효과에 대한 연구자 추가 임상 결과를 공개했다. 이번에 발표한 내용은 지난해 5월 발표한 8주 간의 연구자 임상에 참여했던 인슐린 의존성 당뇨병 반려견을 대상으로 1년 연장 투약에 대한 장기 안전성을 추가로 검증한 연구 결과다. 연구에서 인슐린과 DWP16001을 1년 동안 1일 1회 또는 3일 1회 병용투여하고, 각 군에게 혈중 케톤 및 젖산탈수소효소(Lactate dehydrogenase, LDH) 검사, 일반 혈액(Complete Blood Cell, CBC) 검사, 혈청화학(serum chemistry) 검사, 전해질 검사, 요검사 등을 시행했다. 혈중 케톤 및 LDH 검사는 ‘당뇨병성 케톤산증’ 평가 검사로 저혈당과 함께 가장 주요한 부작용 평가 지표로 알려졌다. 검사 결과 약물을 투여한 1년 동안 혈중 케톤 및 LDH의 유의적인 변화가 없었다. 당뇨 반려견 치료 및 관리에서 가장 중요한 부작용인 저혈당증 및 당뇨병성 케톤산증은 없는 것으로 확인됐다. 백혈구, 적혈구, 혈소판 및 주요 장기(간, 신장 등)에 대한 유의한 수치 변화는 확인되지 않았다. 대웅제약은 “당뇨 반려견 대상 인슐린 및 DWP16001 1년 장기 투약에 대한 장기 안전성을 확보하게 됐다”라고 평가했다. 이번 연구의 책임 연구자인 윤화영 서울대학교 수의과대학 교수는 “당뇨병 반려견에 인슐린 및 DWP16001 투약을 1년에 걸쳐 장기 적용해 본 결과, 안전성 및 혈당조절효과가 확인됐다”고 말했다. 대웅제약은 두 차례의 연구자 주도임상으로 유효성 및 안전성을 확인한 만큼 실제 동물의약품 출시로 이어질 수 있도록 개발을 가속화 한다는 계획이다. 전승호 대웅제약 대표는 "두 건의 연구자 임상으로 당뇨 반려견에서 DWP16001의 유효성 및 안전성을 모두 확인했다"며 "반려동물 대상 의약품으로 개발해 경구 치료제가 없는 반려동물 시장에서 새로운 선택지를 제시할 수 있을 것으로 기대한다"고 말했다.2022-11-22 09:45:41천승현 -
 1년 넘게 감감...원샷 망막치료제 럭스터나 급여등재될까[데일리팜=어윤호 기자] 원샷 유전자치료제 '럭스터나'의 보장성 확대 논의가 지지부진하다. 한국노바티스의 유전성망막질환(IRD, Inherited Retinal Dystrophy)치료제 럭스터나(보레티진 네파보벡)가 연내 건강보험심사평가원 약제급여평가위원회에 상정될 수 있을지 주목된다. 이 약은 지난해 9월 급여 신청을 제출했지만 아직까지 약제급여기준소위 통과 소식이 들리지 않고 있다. 다만 향후 절차 진행 속도에 따라 내년 상반기 등재도 기대할 수 있는 상황이다. 럭스터나는 IRD 발생 원인 중 하나인 결핍, 결함이 있는 RPE65 유전자를 단 1회 투여만으로 정상 유전자로 대체해 기능을 회복시킨다. 질병의 근본적인 치료가 가능한 셈이다. 이 약은 미국 FDA가 2014년 혁신적 치료제(Breakthrough Therapy), 2016년 희귀의약품(Orphan Drug), 2017년 우선 심사(Priority Review) 대상으로 지정하며 2017년 신속 승인을 획득하기도 했다. IRD는 망막 시세포 구조와 기능을 담당하는 유전자에 돌연변이가 생겨 시각 손실이 발생하는 희귀 난치성 질환이다. 약 20가지 이상 다양한 안과 질환을 포함하며 300여개 원인 유전자가 있다. RPE65 유전자 변이로 인하여 발생하는 IRD는 눈에 들어온 시각 정보를 신경 신호로 변환하고 뇌로 전달하는 망막 내 시각 회로(visual cycle)에 이상이 생긴다. RPE65 유전자 돌연변이로 시각 회로에 필수적인 RPE65 단백질이 감소, 망막세포가 파괴되면서 시야가 점차 좁아지다가 결국 실명에까지 이를 수 있다. 한편 럭스터나는 RPE65 유전자의 이중대립형질 돌연변이가 확인된 유전성 망막 질환 환자를 대상으로 진행된 3상 임상을 통해 유효성을 입증했다. 임상 결과, 치료 1년 시점에 럭스터나 치료를 받은 환자군의 시기능(Functional Vision)이 치료를 받지 않은 대조군보다 통계적으로 유의하게 개선됐다. 일상적인 보행 환경을 재현해 다양한 조도에서 여러 가지 높이의 장애물 코스를 통과하는 능력을 평가하는 '다중 휘도 운동성 검사(MLMT, Multi-Luminance Mobility Test)'의 평균 점수를 1차 평가변수로 치료 1년 시점에 평가한 결과, 럭스터나 치료군의 점수 변화는 1.8점으로, 대조군의 점수 변화인 0.2점보다 1.6점 높았다.2022-11-22 06:00:21어윤호
1년 넘게 감감...원샷 망막치료제 럭스터나 급여등재될까[데일리팜=어윤호 기자] 원샷 유전자치료제 '럭스터나'의 보장성 확대 논의가 지지부진하다. 한국노바티스의 유전성망막질환(IRD, Inherited Retinal Dystrophy)치료제 럭스터나(보레티진 네파보벡)가 연내 건강보험심사평가원 약제급여평가위원회에 상정될 수 있을지 주목된다. 이 약은 지난해 9월 급여 신청을 제출했지만 아직까지 약제급여기준소위 통과 소식이 들리지 않고 있다. 다만 향후 절차 진행 속도에 따라 내년 상반기 등재도 기대할 수 있는 상황이다. 럭스터나는 IRD 발생 원인 중 하나인 결핍, 결함이 있는 RPE65 유전자를 단 1회 투여만으로 정상 유전자로 대체해 기능을 회복시킨다. 질병의 근본적인 치료가 가능한 셈이다. 이 약은 미국 FDA가 2014년 혁신적 치료제(Breakthrough Therapy), 2016년 희귀의약품(Orphan Drug), 2017년 우선 심사(Priority Review) 대상으로 지정하며 2017년 신속 승인을 획득하기도 했다. IRD는 망막 시세포 구조와 기능을 담당하는 유전자에 돌연변이가 생겨 시각 손실이 발생하는 희귀 난치성 질환이다. 약 20가지 이상 다양한 안과 질환을 포함하며 300여개 원인 유전자가 있다. RPE65 유전자 변이로 인하여 발생하는 IRD는 눈에 들어온 시각 정보를 신경 신호로 변환하고 뇌로 전달하는 망막 내 시각 회로(visual cycle)에 이상이 생긴다. RPE65 유전자 돌연변이로 시각 회로에 필수적인 RPE65 단백질이 감소, 망막세포가 파괴되면서 시야가 점차 좁아지다가 결국 실명에까지 이를 수 있다. 한편 럭스터나는 RPE65 유전자의 이중대립형질 돌연변이가 확인된 유전성 망막 질환 환자를 대상으로 진행된 3상 임상을 통해 유효성을 입증했다. 임상 결과, 치료 1년 시점에 럭스터나 치료를 받은 환자군의 시기능(Functional Vision)이 치료를 받지 않은 대조군보다 통계적으로 유의하게 개선됐다. 일상적인 보행 환경을 재현해 다양한 조도에서 여러 가지 높이의 장애물 코스를 통과하는 능력을 평가하는 '다중 휘도 운동성 검사(MLMT, Multi-Luminance Mobility Test)'의 평균 점수를 1차 평가변수로 치료 1년 시점에 평가한 결과, 럭스터나 치료군의 점수 변화는 1.8점으로, 대조군의 점수 변화인 0.2점보다 1.6점 높았다.2022-11-22 06:00:21어윤호 -
 목암연구소·차백신연구소, AI 신약개발 공동연구 MOU[데일리팜=김진구 기자] 목암생명과학연구소는 차백신연구소와 'AI(인공지능) 기반 신약 후보물질 발굴을 위한 상호협력 업무협약'을 체결했다고 21일 밝혔다. 이번 협약을 통해 양사는 각종 AI 알고리즘을 이용한 새로운 신약 개발을 위해 상호 협력키로 했다. AI를 활용한 신약물질 발굴·개선을 위한 공동연구, 신약 후보물질 개발의 사전 준비 협의를 위한 물적& 8729;인적자원 교류 협력을 진행한다. 지식재산권 출원과 논문 발표에도 힘을 모은다. 목암연구소는 자체 보유한 AI 알고리즘을 통해 해당 데이터를 분석해 세포와 신호물질 전달과정에서 발생하는 기전에 대한 연구를 담당한다. 차백신연구소는 자체 보유한 획기적인 면역증강 플랫폼을 기반으로 확보한 연구 데이터를 제공한다. 목암연구소는 이번 협약으로 백신 개발 분야에서 인공지능 기술의 효용성을 입증하고, 이를 바이오 신약 개발로 확장하도록 연구 기반을 다질 계획이다. 김선 목암생명과학연구소장은 "목암연구소의 연구 역량에 차백신연구소의 최신 제조 기술을 더해 신규 백신 개발과 인공지능 신약 개발의 새로운 패러다임을 만들고자 한다"고 말했다. 염정선 차백신연구소 대표는 "신약 개발은 막대한 비용과 기간이 소요되는 만큼, 이를 단축하기 위한 AI를 활용한 움직임이 활발히 전개되고 있다"며 "독자개발한 면역증강 플랫폼을 기반으로 프리미엄 백신을 개발 중인 차백신연구소의 기술력에 목암생명과학연구소의 AI 기술력을 접목해 추가적인 신약 개발을 적극 추진하겠다"고 말했다. 목암생명과학연구소는 AI 기반 신약개발 연구소로 탈바꿈하기 위해 지난 1월 서울대학교 AI연구원과 신약 연구 플랫폼을 구축하는 공동연구 협약을 체결했다. 이를 통해 유전체 연구, mRNA 플랫폼 연구, 희귀질환 연구 등 혁신신약 연구·개발에 힘쓰고 있다.2022-11-21 09:25:45김진구
목암연구소·차백신연구소, AI 신약개발 공동연구 MOU[데일리팜=김진구 기자] 목암생명과학연구소는 차백신연구소와 'AI(인공지능) 기반 신약 후보물질 발굴을 위한 상호협력 업무협약'을 체결했다고 21일 밝혔다. 이번 협약을 통해 양사는 각종 AI 알고리즘을 이용한 새로운 신약 개발을 위해 상호 협력키로 했다. AI를 활용한 신약물질 발굴·개선을 위한 공동연구, 신약 후보물질 개발의 사전 준비 협의를 위한 물적& 8729;인적자원 교류 협력을 진행한다. 지식재산권 출원과 논문 발표에도 힘을 모은다. 목암연구소는 자체 보유한 AI 알고리즘을 통해 해당 데이터를 분석해 세포와 신호물질 전달과정에서 발생하는 기전에 대한 연구를 담당한다. 차백신연구소는 자체 보유한 획기적인 면역증강 플랫폼을 기반으로 확보한 연구 데이터를 제공한다. 목암연구소는 이번 협약으로 백신 개발 분야에서 인공지능 기술의 효용성을 입증하고, 이를 바이오 신약 개발로 확장하도록 연구 기반을 다질 계획이다. 김선 목암생명과학연구소장은 "목암연구소의 연구 역량에 차백신연구소의 최신 제조 기술을 더해 신규 백신 개발과 인공지능 신약 개발의 새로운 패러다임을 만들고자 한다"고 말했다. 염정선 차백신연구소 대표는 "신약 개발은 막대한 비용과 기간이 소요되는 만큼, 이를 단축하기 위한 AI를 활용한 움직임이 활발히 전개되고 있다"며 "독자개발한 면역증강 플랫폼을 기반으로 프리미엄 백신을 개발 중인 차백신연구소의 기술력에 목암생명과학연구소의 AI 기술력을 접목해 추가적인 신약 개발을 적극 추진하겠다"고 말했다. 목암생명과학연구소는 AI 기반 신약개발 연구소로 탈바꿈하기 위해 지난 1월 서울대학교 AI연구원과 신약 연구 플랫폼을 구축하는 공동연구 협약을 체결했다. 이를 통해 유전체 연구, mRNA 플랫폼 연구, 희귀질환 연구 등 혁신신약 연구·개발에 힘쓰고 있다.2022-11-21 09:25:45김진구 -
 JW중외제약, 새 기전 탈모치료제 전임상 결과 공개[데일리팜=김진구 기자] JW중외제약은 Wnt 표적 기전의 탈모치료제 'JW0061'의 전임상 효능평가 결과를 지난 15~20일 일본에서 열린 'Wnt 2022' 학회에서 최초 공개했다고 21일 밝혔다. 이 후보물질은 피부와 모낭 줄기세포에 있는 Wnt 신호전달경로를 활성화해 모낭 증식과 모발 재생을 촉진시키는 기전이다. Wnt 신호전달경로는 배아 발생 과정에서 피부 발달과 모낭 형성에 중요한 역할을 한다. 피부 줄기세포가 모낭 줄기세포로 변해 모낭으로 분화하는데 필요하다. 모근 끝에 위치해 모발의 성장과 유지를 조절하는 모유두(Dermal Papilla) 세포 증식에도 관여한다. 전임상에선 JW0061은 모유듀 세포에 있는 GFRA1 단백질에 직접 결합해 Wnt 신호전달경로가 활성화되는 작용기전이 새롭게 확인됐다. 모발이 자라는 생장기의 발모와 모낭 수 증가 효과에 대한 동물실험 결과도 발표했다. JW0061와 표준치료제, 위약을 각 실험부위에 도포한 결과 약물 도포를 시작한 지 34일 시점에서 JW0061이 위약군 대비 모발 성장과 모낭 신생성 효과가 있는 것으로 확인됐다. 표준치료제 대비 동등 이상의 발모 효과를 검증했으며, JW0061과 표준치료제 병용요법은 최대 발모 효과를 냈다. 모발의 성장은 생장기·퇴행기·휴지기 순으로 세 단계가 주기적으로 반복되는데, JW0061은 평균 50일 이상 소요되는 생장기 진입 시점을 15일 이상 앞당긴다. JW중외제약은 앞으로 JW0061을 기존 탈모치료제를 보완·대체하는 새로운 혁신신약으로 개발할 방침이다. 2024년 상반기 임상시험 개시를 목표로 GLP 비임상 독성평가를 하고 있다. 박찬희 JW그룹 최고기술책임자(CTO)는 "현재 시판 중인 안드로겐성 탈모치료제와 임상 물질 중에서 직접적으로 약물과 단백질이 결합하는 타깃 분석과 발모 작용기전을 명확히 규명한 약물은 없다"면서 "이번 JW0061의 연구 결과는 GFRA1을 타깃으로 하는 저분자 약물의 최초 보고 사례로 혁신적인 탈모치료제 후보물질로 주목 받을 것"이라고 말했다. 한편 Wnt 학회는 'Wnt 신호전달' 분야의 전 세계 석학, 연구자들이 최신 지견과 연구 결과를 교류하는 장이다. 올해는 Wnt 단백질이 발견된 지 40년이 되는 해로, 이번 학회는 3년 만에 일본 효고현 아와지 유메부타이에서 대면으로 개최됐다.2022-11-21 09:17:42김진구
JW중외제약, 새 기전 탈모치료제 전임상 결과 공개[데일리팜=김진구 기자] JW중외제약은 Wnt 표적 기전의 탈모치료제 'JW0061'의 전임상 효능평가 결과를 지난 15~20일 일본에서 열린 'Wnt 2022' 학회에서 최초 공개했다고 21일 밝혔다. 이 후보물질은 피부와 모낭 줄기세포에 있는 Wnt 신호전달경로를 활성화해 모낭 증식과 모발 재생을 촉진시키는 기전이다. Wnt 신호전달경로는 배아 발생 과정에서 피부 발달과 모낭 형성에 중요한 역할을 한다. 피부 줄기세포가 모낭 줄기세포로 변해 모낭으로 분화하는데 필요하다. 모근 끝에 위치해 모발의 성장과 유지를 조절하는 모유두(Dermal Papilla) 세포 증식에도 관여한다. 전임상에선 JW0061은 모유듀 세포에 있는 GFRA1 단백질에 직접 결합해 Wnt 신호전달경로가 활성화되는 작용기전이 새롭게 확인됐다. 모발이 자라는 생장기의 발모와 모낭 수 증가 효과에 대한 동물실험 결과도 발표했다. JW0061와 표준치료제, 위약을 각 실험부위에 도포한 결과 약물 도포를 시작한 지 34일 시점에서 JW0061이 위약군 대비 모발 성장과 모낭 신생성 효과가 있는 것으로 확인됐다. 표준치료제 대비 동등 이상의 발모 효과를 검증했으며, JW0061과 표준치료제 병용요법은 최대 발모 효과를 냈다. 모발의 성장은 생장기·퇴행기·휴지기 순으로 세 단계가 주기적으로 반복되는데, JW0061은 평균 50일 이상 소요되는 생장기 진입 시점을 15일 이상 앞당긴다. JW중외제약은 앞으로 JW0061을 기존 탈모치료제를 보완·대체하는 새로운 혁신신약으로 개발할 방침이다. 2024년 상반기 임상시험 개시를 목표로 GLP 비임상 독성평가를 하고 있다. 박찬희 JW그룹 최고기술책임자(CTO)는 "현재 시판 중인 안드로겐성 탈모치료제와 임상 물질 중에서 직접적으로 약물과 단백질이 결합하는 타깃 분석과 발모 작용기전을 명확히 규명한 약물은 없다"면서 "이번 JW0061의 연구 결과는 GFRA1을 타깃으로 하는 저분자 약물의 최초 보고 사례로 혁신적인 탈모치료제 후보물질로 주목 받을 것"이라고 말했다. 한편 Wnt 학회는 'Wnt 신호전달' 분야의 전 세계 석학, 연구자들이 최신 지견과 연구 결과를 교류하는 장이다. 올해는 Wnt 단백질이 발견된 지 40년이 되는 해로, 이번 학회는 3년 만에 일본 효고현 아와지 유메부타이에서 대면으로 개최됐다.2022-11-21 09:17:42김진구
-
 평택공장 제조관리약사 채용(경력무관)
평택공장 제조관리약사 채용(경력무관) -
 환인제약(주) 6월 신입/경력 채용 (~6/14(일))
환인제약(주) 6월 신입/경력 채용 (~6/14(일)) -
 명인제약 【상반기】 경력사원 모집
명인제약 【상반기】 경력사원 모집 -
 가천대길병원 약제부 야간약사(계약직) 채용
가천대길병원 약제부 야간약사(계약직) 채용 -
 국립교통재활병원 정규직 약사 채용공고
국립교통재활병원 정규직 약사 채용공고 -
 야간 계약직(금/일) / 주간 정규직·계약직 약사 공채
야간 계약직(금/일) / 주간 정규직·계약직 약사 공채 -
 일산차병원 약제팀 주간약사 채용
일산차병원 약제팀 주간약사 채용 -
 [SK바이오팜] CNS Research Center 연구원 경력 모집
[SK바이오팜] CNS Research Center 연구원 경력 모집 -
 연세대학교 의료원 신규 약사(정규직) 모집
연세대학교 의료원 신규 약사(정규직) 모집 -
 개발팀 PV/MA/제제연구 부문별 경력채용
개발팀 PV/MA/제제연구 부문별 경력채용 -
 세종 공장 품질관리약사 (3년↑)
세종 공장 품질관리약사 (3년↑) -
 계명대학교 동산의료원 2026학년도 임시직(야간약사) 계약직원 상시 공개채용 공고
계명대학교 동산의료원 2026학년도 임시직(야간약사) 계약직원 상시 공개채용 공고 -
 경상북도 김천의료원 약사 채용
경상북도 김천의료원 약사 채용 -
 [강동성심병원] 약사 채용공고(ASP전담 / 주말 약사)
[강동성심병원] 약사 채용공고(ASP전담 / 주말 약사) -
 [서울 30분] 한림병원 약제센터 야간 약사모집
[서울 30분] 한림병원 약제센터 야간 약사모집 -
 제조관리 약사 채용 신입/경력
제조관리 약사 채용 신입/경력 -
 당진공장 품질관리약사 채용(✨신입우대)
당진공장 품질관리약사 채용(✨신입우대) -
 광주광역시 동구에서 원내약국 약사님을 모십니다~
광주광역시 동구에서 원내약국 약사님을 모십니다~ -
 [광주보훈병원] 정규직 약무직(약사) 공개채용
[광주보훈병원] 정규직 약무직(약사) 공개채용 -
 의정부/노원을지대학교병원 약사 채용
의정부/노원을지대학교병원 약사 채용 -
 해외사업본부 PIS팀 약사 채용(경력무관)
해외사업본부 PIS팀 약사 채용(경력무관) -
 [일산백병원] 약제부 계약직 야간/주말당직약사 (신입/경력) 모집 공고
[일산백병원] 약제부 계약직 야간/주말당직약사 (신입/경력) 모집 공고 -
 약제팀 정규약사(신규/경력) 모집
약제팀 정규약사(신규/경력) 모집



오늘의 TOP 10
- 112.7조 정부 지원금 쏟아진다…K-바이오 R&D 재원 숨통
- 2"감기환자 약국 가고, 진료는 비대면"…ENT, 경영난 심화
- 3실무 깊숙이 침투한 AI…업무 단축 뒤에 숨은 고용 불안
- 4P-CAB 첫 약가유연제 펙수클루...경쟁제품도 신청 만지작
- 52796억 오리지널 인수와 제네릭 매각…보령의 항암제 승부수
- 6도수치료 연 최대 24회 제한…회당 4만원대 관리급여 적용
- 7"AI 오류 책임은 결국 약사에게"…AI기본법 핵심은?
- 8틀린 주민번호로 처방 발행…비대면 진료 허점 노출
- 9겔포스·카네스텐 등 스테디셀러 일반약의 변신과 도전
- 10정부 압박에도 CSO 수수료율 확대 경쟁…시장 사수 몸부림